

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website http://www.jofem.org |
Original Article
Volume 4, Number 4, August 2014, pages 101-109
Autoimmune Polyglandular Syndrome Type II: Epidemiological, Clinical and Immunological Data
Mnif Fatmaa, Elleuch Mounaa, Hajji Raoufb, d, Fourati Hajerc, Masmoudi Hatemc, Abid Mohammeda
aEndocrinology Department, Hedi Chaker University Hospital, Sfax, Tunisia
bInternal Medicine Department, Sidi Bouzid Regional Hospital, Sidi Bouzid, Tunisia
cImmunology Laboratory, Hedi Chaker University Hospital, Sfax, Tunisia
dCorresponding Author: Hajji Raouf, Internal Medicine Department, Sidi Bouzid Regional Hospital, Sidi Bouzid, Tunisia
Manuscript accepted for publication July 29, 2014
Short title: Autoimmune Polyglandular Syndrome
doi: https://doi.org/10.14740/jem227w
| Abstract | ▴Top |
Background: Autoimmune polyglandular syndrome (APS) is characterized by the coexistence of several autoimmune diseases, affecting predominantly the endocrine glands. APS type I is distinguished from type II in which autoimmune thyroiditis, Addison’s disease and diabetes type 1 predominate. This article summarizes extensive epidemiological, clinical, and immunological data of a large population of Tunisian patients with APS II.
Methods: In a retrospective case finding study, we collected data from patients with APS type II who had been treated in our endocrine outpatient clinic between 1980 and 2007. One hundred and six patients fulfilled the criteria of APS II.
Results: Among these patients, 73 were female and 33 were male (mean age 36 and 42 years, respectively; P = 0.04). The mean age at the onset was 38 ± 14.9 years. Of the 106 patients, 76 had hypothyroidism due to autoimmune thyroiditis while 23 had hyperthyroidism (Grave’s disease). Sixty-six patients had type I diabetes mellitus (60.6% females and 39.5% males; P = 0.01). Addison’s disease was diagnosed in 39 patients, primary hypogonadism in 12 cases (11 females and one male), and neuro-hypophysitis was less frequently noted (n = 6). The most frequent coexistence of APS component diseases was between type I diabetes and thyroid disease (56.2%). The time interval between the advent of the first and the second autoimmune endocrinopathies varied considerably with longest time intervals between diabetes type I and thyroid disease and shortest time intervals between Addison’s disease and thyroid disease. Regarding autoantibodies in patients with APS II, anti-thyroid peroxidase antibodies were detected in 67.6%, thyroid stimulating hormone-receptor antibodies in 58%, and anti-thyroglobulin were less frequently positive (43.3%). Type I diabetes-associated antibodies against islet cell (islet cell autoantibodies) and glutamic acid decarboxylase were found in 23.6% and 51.8%, respectively. Antibodies to adrenal cortex were observed in 9.7% and ovarian antibodies in 8.8%.
Conclusion: The present study indicates that most patients with autoimmune thyroid disease will not develop additional endocrine disease. If they do, when a thyroid disease was as a first component disease, the time interval until the onset of a further autoimmune disease was relatively short and when thyroid disease was a second component disease a longer period of time elapsed.
Keywords: Autoimmune polyglandular syndrome; Autoimmunity; Antibody
| Introduction | ▴Top |
Autoimmune polyglandular syndrome (APS) or autoimmune polyendocrine syndrome are rare immune endocrinopathies characterized by the coexistence of at least two endocrine gland diseases that are based on autoimmune mechanisms. Neufeld and Blizzard suggested a classification of APS based on clinical criteria only [1, 2].
APS I, also known as an autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy, is a rare disease affecting young subjects [3, 4]. The highest prevalence has been found in Iranian Jews and Finns [3, 5, 6]. The clinical diagnosis of APS I is made in patients who present with two of the following three cardinal symptoms: chronic candidiasis, acquired hypoparathyroidism, and Addison’s disease [1-3].
Patients develop autoantibodies that correlate with affected tissues and show immune-mediated destruction of endocrine organs. APS is inherited in an autosomal recessive pattern and is now known to be caused by mutations in the autoimmune regulator gene [5-7].
APS II is characterized by Addison’s disease associated with autoimmune thyroid diseases (ATDs) (Schmidt’s syndrome) and/or type I diabetes mellitus (DM) [1, 2]. Compared to type I and II, APS type III does not involve the adrenal cortex [8]. Apart from the absence of adrenal failure, APS type II and III represent a series of genetically influenced diseases that frequently coexist because they share susceptibility genes [9].
APS II is much more common than APS I and occurs in adulthood, mainly in the third or fourth decade, having an estimate prevalence of 1.4 - 2/100,000 in the general population and a higher incidence among females [10].
APS II is commonly associated with additional autoimmune diseases, such as celiac disease, pernicious anemia, chronic hepatitis, hypergonadotropic hypogonadism and vitiligo [10].
APS II is a heterogeneous group of diseases with modes of inheritance that are complex and ill defined. Approximately, half of the patients with APS II have relatives with autoimmune disorders; the most known genetic determinant of susceptibility to APS II represents the human leucocyte antigens (HLAs) region [9-11].
It is highly likely that there is a complex interaction between non-HLA loci and environmental factors [10].
In certain families, individuals from several generations may be affected and the disease transmission is consistent with a dominant mode of inheritance involving one or, at most, a few major genes [9].
The aim of this study is to investigate epidemiological, clinical and immunological data from a large group of patients with APS II.
| Materials and Methods | ▴Top |
In a retrospective case finding study, we collected data from patients with APS type II who had been treated in our endocrine outpatient clinic between 1980 and 2007. One hundred and six Tunisian patients fulfilled the criteria of APS II.
Adrenal insufficiency was confirmed by a low serum cortisol sample drawn between 6:00 and 8:00 am or an unstimulated serum cortisol after cosyntropin test (serum cortisol level less than 200 ng/mL after 30 and/or 60 min) with an increase in serum ACTH levels.
The thyroid abnormalities and DM are diagnosed in the conventional manner: thyrotropin secreting hormone (TSH) and serum-free thyroxin are used to diagnose thyroid disease, and the recommended diagnostic criteria for DM are based on a fasting blood glucose level.
Hypergonadotropic hypogonadism was diagnosed by enhanced follicle stimulating hormone and low testosterone in men and low estradiol levels in women.
The diagnosis of autoimmune hypophysitis was made after carefully exhaustive analyses of other causes of pituitary gland damage.
All hormone levels were measured with radioimmunoassay or immune-radiometric assay. The loading tests of the pituitary hormones to the respective hypothalamic stimulating hormones were performed when necessary.
ELISA was applied for autoantibodies against thyroid peroxydase (TPO; positive > 100 UI/mL), thyroglobulin (Tg; positive > 70 UI/mL), TSH receptor (positive > 2 UI/mL) and glutamic acid decarboxylase (GAD; positive > 10 UI/mL). Indirect immunofluorescence was used for adrenal cortex, ovarian, islet cell autoantibodies (ICAs), and antinuclear (AAN).
Statistical analysis
Group comparisons were performed by the use of Chi-deux-test for qualitative variables and Student’s t-test for quantitative variables. ANOVA test compared several variables. Statistic software was used: SPSS (version 13), R 2.6.0 (free), NTSYSpc 2.1, SPAD (version 4.5), and Matlab (version 7). Statistical significance was set at P value less than 0.05.
| Results | ▴Top |
One hundred and six patients with APS II have been followed regularly in our endocrine outpatient clinic since 1980. Seventy-three were females and 33 were males, which corresponds to 2.21 female to male ratio. The mean age of occurrence is 38 ± 14.9 years. The incidence of new cases has one peak during 20 - 30 years and a larger peak in the fourth and fifth decades. The age when our patients develop APS II is earlier in females (mean age 36 years) than in males (mean age 42 years) with P of 0.04. A family history of autoimmune diseases was noted in 43.3%, with highest frequency of ATDs of 31.1%.
The analysis of clinical data showed that ATD had the highest prevalence (n = 99; 93.4%), with an unequal number of Hashimoto thyroiditis (HT) (n = 76; 76.7%) and Grave’s disease (n = 23; 23.2%).
Type I DM, in the second position, was observed in 66 patients. Addison’s disease was noted in 39 cases (36.8%), whereas hypergonadotropic hypogonadism (n = 12; 11.3 %) and neuro-hypophysitis (n = 6; 5.6%) occurred less frequently (Fig. 1A).
Click for large image | Figure 1. (A) Frequency of autoimmune endocrine diseases in 106 patients with APS II. (B) Frequency of autoimmune non-endocrine diseases in 106 patients with APS II. |
With regard to non-endocrine autoimmune component diseases, vitiligo (n = 6; 5.6%) was most frequently observed, followed by alopecia areata (n = 4; 3.7%) and Sjogren’s syndrome (n = 3; 2.8%). The other autoimmune diseases occurred less frequently (Fig. 1B).
Type I DM was the first component disease of APS in 47.1% of cases, whereas thyroid disease was objected in 44% and Addison’s disease in 24.5% of cases. Concomitant presentation occurred in 25 cases (26%), with most frequent association between thyroid diseases and Addison’s disease (52%).
The most frequent coexistence of APS component diseases was between DT1 and ATD (56.2%). Less commonly, coexistence occurred between thyroid and Addison’s disease (26%); the other associations were less frequent (Fig. 2).
Click for large image | Figure 2. Most frequent combinations of endocrine component diseases in 106 patients with APS II. Only the patients with two diseases were noted. Adrenal: Addison’s disease; Thyroid: autoimmune thyroid disease; Diabetes: type I diabetes; Gonad: gonadal failure; POF: premature ovarian failure; Pern.A: pernicious anemia; GJS: Sjogren’s syndrome; Celiac: celiac disease; AH: autoimmune hepatitis; AT: autoimmune thrombocytopenia. |
The time interval between the manifestation of the first and second autoimmune endocrinopathies varied considerably (Fig. 3), with the longest time interval between type I diabetes and thyroid disease (127 months = 10.3 years), but a shorter time interval between Addison’s disease and thyroid disease (17.6 months = 1.4 years).
Click for large image | Figure 3. Time interval between manifestation of first (abscissa) and second endocrine component diseases (ordinate) in 106 patients with APS II. Medians, first and third quartiles as well as ranges of time (months) between manifestation of first and further diseases are shown. |
In general, when a thyroid disease was present as a first component disease, the time interval until the onset of a further endocrinopathy was relatively short (4.3 years between thyroid/diabetes and 1.4 years between adrenal/diabetes with non-significant differences between these time intervals). On the other hand, when thyroid disease was the second component disease, a longer period of time elapsed (10.3 years between diabetes/thyroid and 5.3 years between adrenal/thyroid with non-significant differences between these time intervals). The difference between time intervals in thyroid/other endocrinopathy (30 months = 2.5 years) and other endocrinopathy/thyroid (122 months = 10.1 years) is statistically significant (P = 0.00003).
Regarding autoantibodies in patients with APS II, antibodies against TPO were the most frequent (67.6%), antibodies against TSH-R (58%), antibodies against Tg were less frequent (43.3%). Type I diabetes-associated antibodies directed against GAD were found in 51.8 % and ICA in only 23.6%. Other autoantibodies were less frequent: anti-adrenal cortex were positive in 9.7% and anti-ovarian in 8.8% of cases.
ATDs in APS II
ATDs were present in 99 patients, the mean age of occurrence was 38 years and the trend of presentation is shown in Figure 4. HT had the highest prevalence (n = 45 with goiter and n = 31 without goiter) whereas Grave’s disease was noted in 23 cases.
Click for large image | Figure 4. Age at manifestation of the three main component autoimmune endocrinopathies in 106 patients with APS II. |
Grave’s disease was manifested at an earlier age (mean age: 33 years), goitrous HT (mean age: 35 years) and atrophic HT later (mean age: 46 years); the difference between ages is statistically different.
The female/male ratio was 2.8. No significant male to female preponderances were noted for ATD and their different component.
Anti-TPO and/or anti-Tg antibodies were detected in 74.6% of patients with ATD with a higher level of antibodies in HT (mean level of anti-TPO: 1,700 UI/mL; anti-Tg: 1,600 UI/mL).
Type I DM
Among our patients with APS II, type I DM was present in 63% of cases. Type I DM was manifested earlier, and the mean age of presentation of DM was 31 years.
Significant female preponderances were noted for type I DM (P = 0.01). The female/male ratio was 1.53. Pancreatic-autoantibodies (ICA and/or anti-GAD) were positive in 55% of cases. Thirty-one percent of patients presented with a non-acute onset of diabetes and insulin therapy was instaurated after a mean duration of 4.7 years in these cases.
Addison’s disease
Among our 106 patients, 39 had Addison’s disease. The mean age at presentation was 39 years and the trend of presentation is reported in Figure 4.
Women are affected three times more often than men; the female/male ratio was 3.1. Adrenal crisis revealed the disease in 20.5% of cases. The antibodies of anti-adrenal cortex were positive only in 21.8% of cases because they were analyzed many years after the disease diagnosis.
Cluster analysis
Through the examination of various patient characteristics, clinical subgroups of patients with APS II have been identified. Using a number of cluster analysis approaches in the examination of patients, we were able to identify four groups or “classes” of patients (Fig. 5): 1) Class I: the component of this group was manifested at an early age (mean age: 28 years), and it corresponded to patients who had frequently type I DM associated with Addison’s disease. 2) Class II: corresponded to females, whose mean age at presentation was 29 years, and presented goitrous HT and POF. 3) Class III: corresponded to adults whose mean age at presentation was 43 years, who had atrophic HT and Addison’s disease. 4) Class IV: corresponded to adult females whose mean age at presentation was 38 years, with the frequent association of type I DM and HT.
Click for large image | Figure 5. The result of cluster analysis showing clinical subgroups of patients with APS II. |
We concluded that if a young patient developed an Addison’s disease, he would have type I DM associated, but if he was an adult he would develop atrophic HT associated with Addison’s disease.
If a young woman had a HT, she would develop a POF, but if she was adult she would develop a type I DM.
| Discussion | ▴Top |
APS has become an increasingly recognized clinical entity in endocrinology, joining a growing list of disorders likely to be mediated by autoimmune pathogenic mechanisms.
There is controversy over the syndrome classification. According to Neufeld [1, 2], APS II refers to Addison’s disease and thyroid autoimmunity or type I DM; APS III refers to thyroid autoimmunity and another autoimmunity (but not Addison’s disease). Others authors, with whom we tend to agree, consider all the above combinations in APS II [9].
Muir [9], considering that a distinction between APS II and III is arbitrary, suggested that APS II and III represent a series of genetically influenced diseases that frequently coexist because they share susceptibility genes.
This study reports epidemiological, clinical and serological data of 106 patients with APS II. APS II is rather rare with the incidence of 1.4 - 4.5 cases/100,000 inhabitants [10]. The disease affected mainly adult women; the mean age at presentation is 35 years [10, 12, 13]. The female/male ratio is 2.7 - 3.7 [10, 12, 13]. In our study the mean age at presentation was 38 years with female/male ratio 2.21. Significant male and female preponderances were noted for type I DM. Dittmar and Kahaly [12] noted a significant difference between males and females in type I DM and thyroiditis but he did not find a difference for manifest Graves’s disease and Addison’s diseases.
Familial aggregation of APS II and III does not seem to be inherited as Mendelian traits [9, 10, 14, 15]. In certain families, individuals from several generations may be affected and the disease transmission is consistent with a dominant mode of inheritance. Therefore, APS II appears to follow an autosomal dominant mode of inheritance with variable penetrance [9, 14, 15]. In our study, a family history of autoimmune diseases was noted in 43.3%, with highest frequency of ATDs (31.1%). Because of a 50% transmission rate from affected mothers to daughters, the production of anti-Tg and anti-TPO antibodies is considered to be an autosomal dominant gene [9, 10].
The most prevalent endocrinologic abnormality is ATD, occurring from 65.5% to 97% of APS II, followed by type I DM, which are reported to develop in 35% to 70%, Addison’s, being less frequent, occurred in 18.5% to 23% [10, 11, 13]. Other minor autoimmune disorders including gonadal failure and hypophysitis pernicious anemia occurred with different rates (Table 1).
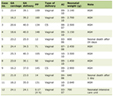 Click to view | Table 1. Prevalence of Component Disease in APS II |
Compared with the studies in literature (Table 1), the present study about patients with APS II found similar prevalence of thyroid disease as in Wyermann’s study [13], and similar prevalence of type I DM as in Dittmar’s [12] but a higher prevalence of Addison’s disease, hypophysitis and celiac disease. A lower prevalence of vitiligo and pernicious anemia was also noted. The prevalence of gonadal failure and alopecia areata was variable in different series.
Epidemiological data showed varying time intervals between the manifestations of different component diseases of APS II, with longest time interval between type I DM and thyroid disease and shortest time between Addison’s and thyroid disease.
Dittmar and Kahaly [12] also noted in a study of 151 patients with APS II that the longest time intervals were between type I DM and thyroid disease (13.3 ± 11.8 years) and between vitiligo and thyroid disease (16.3 ±13.3 years), and the shortest time interval was between Addison’s disease and thyroid disease. Thus, time intervals between first and second component disease manifestations were significantly different, where thyroid disease and vitiligo, respectively, were the first disease component.
In the APS II series of Neufeld et al [1, 2] and in patients with type I DM, thyroid disease was the autoimmune condition with a later age of onset. Thus, polyglandular involvement among the total population of patients with ATD is infrequent and when it does occur, the onset of other APS components has often preceded the diagnosis of thyroid disease.
Irvine [15] reported that 69% of patients with autoimmune adrenalitis have concomitant ATD, with Grave’s disease and primary atrophic hypothyroidism being more common than goitrous autoimmune thyroiditis.
In our studies, 48.7% of patients with Addison’s disease have concomitant autoimmune disease, with primary atrophic hypothyroidism in 55.5% and goitrous thyroiditis in 33.3%.
The results suggest that early screening of patients with autoimmune endocrine diseases enhances the possibility of identifying patients at risk for APS II [10, 15].
Discrepancies may occur between the clinical expression of APS and the presence or absence of positive results of organ-specific serologic testing. Thus, positive autoimmune serologic results may precede the onset of clinical disease [10].
Refined endocrine function tests can now be performed in the follow-up of patients suspected to have APS, and first-degree relatives could be screened every few years with standard tests for glycemia and for early or partial deficiency, which may indicate future glandular failure [10, 14, 16].
The frequencies of the relevant autoantibodies detectable at the clinical onset of the diseases constituting APS II are summarized in Table 2. Autoantibodies against TPO and Tg are the more frequent positive in APS. This is a positive correlation with the frequency of ATD which represents the most component of APS II [10, 11, 13]. Weyermann et al [13] noted a high percentage (70%) of his patients had high titers of antiparietal cell antibodies (APC). The clinical importance of APC is demonstrated by the associations of autoimmune gastropathy such as pernicious anemia, gastritis, and iron deficiency anemia. Christophe et al [17] noted that type I DM has a high prevalence of PCA (20.9%), and that anti-TPO were more frequent in PCA-positive patients than in those without PCA suggesting an association between gastric and thyroid autoimmunity.
Click to view | Table 2. Prevalence of Positive Autoantibodies in APS II |
Patients with polyglandular autoimmunity also have been reported to have an increased incidence of circulating non-organ-specific autoantibodies, including autoantibodies against mitochondria, single-stranded deoxyribonucleic acid and double-stranded deoxyribonucleic acid and ribonucleic acid [18].
Circulating immune complexes have been documented in patients with HT, Grave’s disease and insulin-dependent DM [19, 20]. In our study, circulating immune complexes, cryoglobulinemia and AAN antibodies were found in 23.8%, 14.2% and 13.5% of cases respectively.
HLAs play a role in conditioning T lymphocyte response to antigens, and the association of different HLA alleles with many autoimmune disorders has been shown [9, 21]. HLA-D subgroup DR3 and HLA-B8 have been found to be associated with APS II [22].
A degree of genetic polymorphism in the expression of the HLA-DR antigen subgroup may also account for this variability among families in the expression of APS II [12, 23, 24].
The role of HLA in APS II and its component diseases has been extensively examined using case-control association studies. Susceptibility is usually associated with the DR3DQB1*0201 and DR4DQB1*0302 haplotypes, particularly heterozygotes for the two haplotypes [9, 24, 25].
CTLA4 on chromosome 2 is a candidate gene for autoimmune diseases because of its important role in the T cell proliferate response. This locus has been related to type I DM and ATD, Grave’s disease and HT [21, 26]. CTLA4 gene may be one of the susceptible genes that can influence the onset of APS II and its component diseases.
Conclusion
A continued suspicion of other glandular hypofunction should be maintained in following patients with any type of endocrine gland hypofunction, since the risk of multiple glandular involvements is significant.
Autoantibodies are useful markers for the prediction of the development of APS.
In view of the possible long time interval between manifestation of the first and further autoimmune endocrinopathies, regular and long-term examination of patients with endocrine autoimmune disorders seems necessary.
Conflict of Interests
We declare no conflict of interests.
| References | ▴Top |
- Neufeld M, Blizzard RM. Polyglandular autoimmune disease. In: Pinchera A, Doniach D, Fenzi GF, Baschieri L, eds. Symposium on autoimmune aspects of endocrine disorders. New York: Academic Press; 1980:357-365.
- Neufeld M, Maclaren NK, Blizzard RM. Two types of autoimmune Addison's disease associated with different polyglandular autoimmune (PGA) syndromes. Medicine (Baltimore). 1981;60(5):355-362.
doi - Ahonen P, Myllarniemi S, Sipila I, Perheentupa J. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med. 1990;322(26):1829-1836.
doi pubmed - Ahonen P. Autoimmune polyendocrinopathy—candidosis—ectodermal dystrophy (APECED): autosomal recessive inheritance. Clin Genet. 1985;27(6):535-542.
doi pubmed - Betterle C, Greggio NA, Volpato M. Clinical review 93: Autoimmune polyglandular syndrome type 1. J Clin Endocrinol Metab. 1998;83(4):1049-1055.
doi pubmed - Zlotogora J, Shapiro MS. Polyglandular autoimmune syndrome type I among Iranian Jews. J Med Genet. 1992;29(11):824-826.
doi - Blizzard RM, Tomasi TB, Christy NP. Autoantibodies against Thyroid and Adrenal Tissue in a Patient with Multiple, Primary Endocrine Deficiencies. J Clin Endocrinol Metab. 1963;23:1179-1180.
doi pubmed - Majeroni BA, Patel P. Autoimmune polyglandular syndrome, type II. Am Fam Physician. 2007;75(5):667-670.
pubmed - Muir A, She JX. Advances in the genetics and immunology of autoimmune polyglandular syndrome II/III and their clinical applications. Ann Med Interne (Paris). 1999;150(4):301-312.
- Betterle C, Zanchetta R. Update on autoimmune polyendocrine syndromes (APS). Acta Biomed. 2003;74(1):9-33.
pubmed - Phillips DI, Shields DC, Dugoujon JM, Prentice L, McGuffin P, Ree Smith B. Complex segregation analysis of thyroid autoantibodies: are they inherited as an autosomal dominant trait? Hum Hered. 1993;43(3):141-146.
doi pubmed - Dittmar M, Kahaly GJ. Polyglandular autoimmune syndromes: immunogenetics and long-term follow-up. J Clin Endocrinol Metab. 2003;88(7):2983-2992.
doi pubmed - Weyermann D, Spinas G, Roth S, Guglielmetti M, Viollier E, Staub JJ. [Combined endocrine autoimmune syndrome—incidence, forms of manifestation and clinical significance]. Schweiz Med Wochenschr. 1994;124(44):1971-1975.
pubmed - Maugendre D, Massart C, Karacatsanis C, Guilhem I, Poirier JY, Sonnet E, Allannic H. Increased prevalence of thyroid autoantibodies and subclinical thyroid failure in relatives of patients with overt endocrine disease-associated diabetes but not type 1 diabetes alone. Diabetes Metab. 1997;23(4):302-307.
pubmed - Irvine WJ. Autoimmunity in endocrine disorders. In: Besser GH, ed. Advenced medicine: proceedings of 13th annual symposium of advanced medicine 1977. Woodstock, NY: Beekman Pubs; 1977:115-140.
- Brix TH, Hansen PS, Kyvik KO, Hegedus L. Aggregation of thyroid autoantibodies in first-degree relatives of patients with autoimmune thyroid disease is mainly due to genes: a twin study. Clin Endocrinol (Oxf). 2004;60(3):329-334.
doi - De Block CE, De Leeuw IH, Van Gaal LF. High prevalence of manifestations of gastric autoimmunity in parietal cell antibody-positive type 1 (insulin-dependent) diabetic patients. The Belgian Diabetes Registry. J Clin Endocrinol Metab. 1999;84(11):4062-4067.
pubmed - Trence DL, Morley JE, Handwerger BS. Polyglandular autoimmune syndromes. Am J Med. 1984;77(1):107-116.
doi - Baethge BA, Levine SN, Wolf RE. Antibodies to nuclear antigens in Graves' disease. J Clin Endocrinol Metab. 1988;66(3):485-488.
doi pubmed - Eberhard BA, Laxer RM, Eddy AA, Silverman ED. Presence of thyroid abnormalities in children with systemic lupus erythematosus. J Pediatr. 1991;119(2):277-279.
doi - Caillat-Zucman S. Genetic predisposition to autoimmune endocrine diseases. Ann Med Interne (Paris). 1999;150(3):221-234.
- Hrda P, Sterzl I, Matucha P, Korioth F, Kromminga A. HLA antigen expression in autoimmune endocrinopathies. Physiol Res. 2004;53(2):191-197.
pubmed - Albergoni P, Gazzola MV, Slanzi E, Carcassi C, Dal Pra C, Moscon A, Betterle R. HLA DR and DQ association with autoimmune Addison's disease in Italian patients. Genes Immunity. 2003;4:535.
- Huang W, Connor E, Rosa TD, Muir A, Schatz D, Silverstein J, Crockett S, et al. Although DR3-DQB1*0201 may be associated with multiple component diseases of the autoimmune polyglandular syndromes, the human leukocyte antigen DR4-DQB1*0302 haplotype is implicated only in beta-cell autoimmunity. J Clin Endocrinol Metab. 1996;81(7):2559-2563.
pubmed - Myhre AG, Undlien DE, Lovas K, Uhlving S, Nedrebo BG, Fougner KJ, Trovik T, et al. Autoimmune adrenocortical failure in Norway autoantibodies and human leukocyte antigen class II associations related to clinical features. J Clin Endocrinol Metab. 2002;87(2):618-623.
doi pubmed - Donner H, Rau H, Walfish PG, Braun J, Siegmund T, Finke R, Herwig J, et al. CTLA4 alanine-17 confers genetic susceptibility to Graves' disease and to type 1 diabetes mellitus. J Clin Endocrinol Metab. 1997;82(1):143-146.
pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.