

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website http://www.jofem.org |
Case Report
Volume 9, Number 5, October 2019, pages 165-170
Metatropic Dysplasia: A Description of a Newborn With Suspected Epiphyseal Dysplasia
Agnieszka Byrwaa, b, Izabela Michalusa, Paulina Adamieckaa, Elzbieta Jakubowska-Pietkiewicza
aDepartment of Paediatrics, Neonate Pathology and Bone Metabolic Disorders of Medical University of Lodz, Lodz, Poland
bCorresponding Author: Agnieszka Byrwa, Department of Paediatrics, Neonate Pathology and Bone Metabolic Disorders of Medical University of Lodz, ul. Sporna 36/50, 91-738 Lodz, Poland
Manuscript submitted August 4, 2019, accepted August 13, 2019
Short title: A Newborn With Suspected Epiphyseal Dysplasia
doi: https://doi.org/10.14740/jem598
| Abstract | ▴Top |
The paper presents a female neonate with a suspected skeletal defect that was eventually diagnosed as metatropic dysplasia. This is the first case of this disease in our clinic. Clinical course of the disease was discussed as well as findings of additional examinations and the differentiation from other diseases with a similar clinical picture. Newborns have a narrow and elongated chest with extremely short limbs, with bones resembling dumbbells. Other problems associated with dysplasia involve the spine (the vertebrae are excessively flattened), and any excessive movement of the cervical spine may lead to spinal cord damage. The chest may be sunken or excessively bowed which may lead to disable full lung expansion that constrains breathing; also joint deformities occur such as contractures limiting movements in the largest joints. In the collected genetic material no TRPV4 mutation was discovered. There are slightly more than 80 cases of this disease described worldwide.
Keywords: Metatropic dysplasia; Neonate; Bone metabolic; Skeletal deformations
| Introduction | ▴Top |
Metatropic dysplasia is a rare skeletal system defect which is characterised by dwarfism and skeletal system lesions. There are slightly more than 80 cases of this disease described worldwide [1]. Newborns have a narrow and elongated chest with extremely short limbs, with bones resembling dumbbells [2]. There are also reports of children born with a remaining coccyx, the structure of which corresponds to cartilage and shrinks over time [3]. Other problems associated with dysplasia involve the spine (the vertebrae are excessively flattened), and any excessive movement of the cervical spine may lead to spinal cord damage. The chest may be sunken or excessively bowed; also joint deformities occur such as contractures limiting movements in the largest joints including shoulder, elbow, hip and knee. Individuals suffering from this disease may develop arthritis already at the early stage of life. Dysplasia may involve a wide range of relatively mild to life-threatening symptoms. In the most severe cases, narrow thorax and spine abnormalities disable full lung expansion that constrains breathing. At present, there are several distinct forms of metatropic dysplasia identified based on disease’s severity. These are non-lethal type with autosomal recessive transmission, non-lethal dominant type and lethal type causing death before or soon after birth, probably inherited in an autosomal recessive pattern. However, all these forms are now considered as part of one single condition with a wide spectrum of overlapping symptoms [1].
The presentation of the below child description is to draw the attention of physicians of different specialities to the importance of a multi-disciplinary approach and close cooperation between several fields of medicine, as well to the severity of problems that parents of children with such a burden have to face.
| Case Report | ▴Top |
A female newborn child on day 4 of life with a suspected skeletal system deficit was admitted to the Clinic of Pediatrics, Neonate Pathology and Bone Metabolic Diseases for further diagnostics and treatment. Medical history and records indicated that the child was born spontaneously from the first pregnancy, first delivery, in week 38 of pregnancy, with body weight of 2,780 g and Apgar score of 2/5/8/8. Immediately after birth the girl required respiratory support. The obstetrics history with a burden of anemia and colonisation of the birth canal with group B streptococcus (GBS) were revealed. The mother was hospitalised in week 25 of pregnancy due to a suspected fetal chondrodysplasia. After birth, multiple developmental defects of the skeletal system were observed in the child and metaphyseal dysplasia was suspected. The disease occurred in the family for the first time. On admission to the clinic the girl was in a general fair and stable condition. The physical examination revealed the yellowing of the skin, white tongue, excessive hair in the lumbar area, hemangioma on the lids, greyness around the lips, multiple crackles over the lung fields. Moreover, multiple musculoskeletal system defects were present: disproportion between the cerebral and facial cranium, depressed nasal bridge, overlapping skull bones, narrow disproportional thorax, broadened rib endings, deformed spine, protruding spinous processes, additional vertebrae as coccygeal segment, broadened long bone metaphyses, contractures in joints, long fingers and toes. On the first days of her stay at the clinic antibiotic treatment was used due to clinical and radiological symptoms of infection. Temporary saturation falls were also observed in the child, and that decreased after the cervical spine was stabilised with a cervical collar. The girl was fed through a feeding tube; and attempts were made to feed her with a soother however she was only able to eat small amounts by herself. Due to an increasing hypercapnic respiratory failure of up to 166 mm Hg the child was transferred to the intensive care unit (ICU) for further treatment. The girl was intubated and synchronized intermittent mechanical ventilation (SIMV) was initiated.
During hospitalisation imaging examinations of bones of both lower limbs and skull were made. The performed lateral roentgenogram of lower limbs revealed short lower limb and thigh bones with significant metaphyseal flaring, significantly broadened proximal tibial metaphyses with emphasis of the anterior outline, especially the right one, altered shape and structure of uneven outlines of ossification centres for ankle and heel bones (Fig. 1). The picture seemed to support thanatophoric or spondylometaphyseal dysplasia. X-ray of the skull revealed a skull deformity with a large biparietal diameter and a reduced anterior-posterior (AP) diameter, flattened upper part of the skullcap, especially on the left, thin parietal bones, mildly calcified, prominent frontal bone, moreover low-set ears and saddle nose deformity. No pathological intracranial calcifications were observed. Also atypical placement of dens axis was observed, with a possible subluxation, therefore transfontanelle ultrasound was performed, followed by a computed tomography (CT) of the head. In the ultrasound, a smaller posterior cranial fossa was observed, and dilated frontal horns, rounded. CT of the head revealed the atlantodental interval of approximately 3 mm, narrow AP dimension of the spinal canal at the level of C1-C2 of approximately 4.5 mm, depending on the shortening of the posterior C1 arch, the height of cervical and thoracic vertebral bodies significantly decreased, platyspondylia (Fig. 2) only partially ossified; arches and vertebrae well formed, quite massive, ossified. Spinal canal in the cervical segment C3-C7 of approximately 8 mm, in the thoracic segment it is narrow again (4 - 6 mm), especially in the transverse dimension, shortened and too closely located epiphyses of the arches, 12 ribs visible on each side, shortened, especially in the posterior segments which reduces the volume of the thoracic cage (Fig. 3). Lung fields with impaired aeration in dorsal segments of lower lobes bilaterally, and of the left upper lobe; small, shallow fields of atelectasis visible also in the right middle and upper lobes. Due to ambiguous diagnosis of atlantoaxial instability, it was decided to put stabilising collar and nurse with caution.
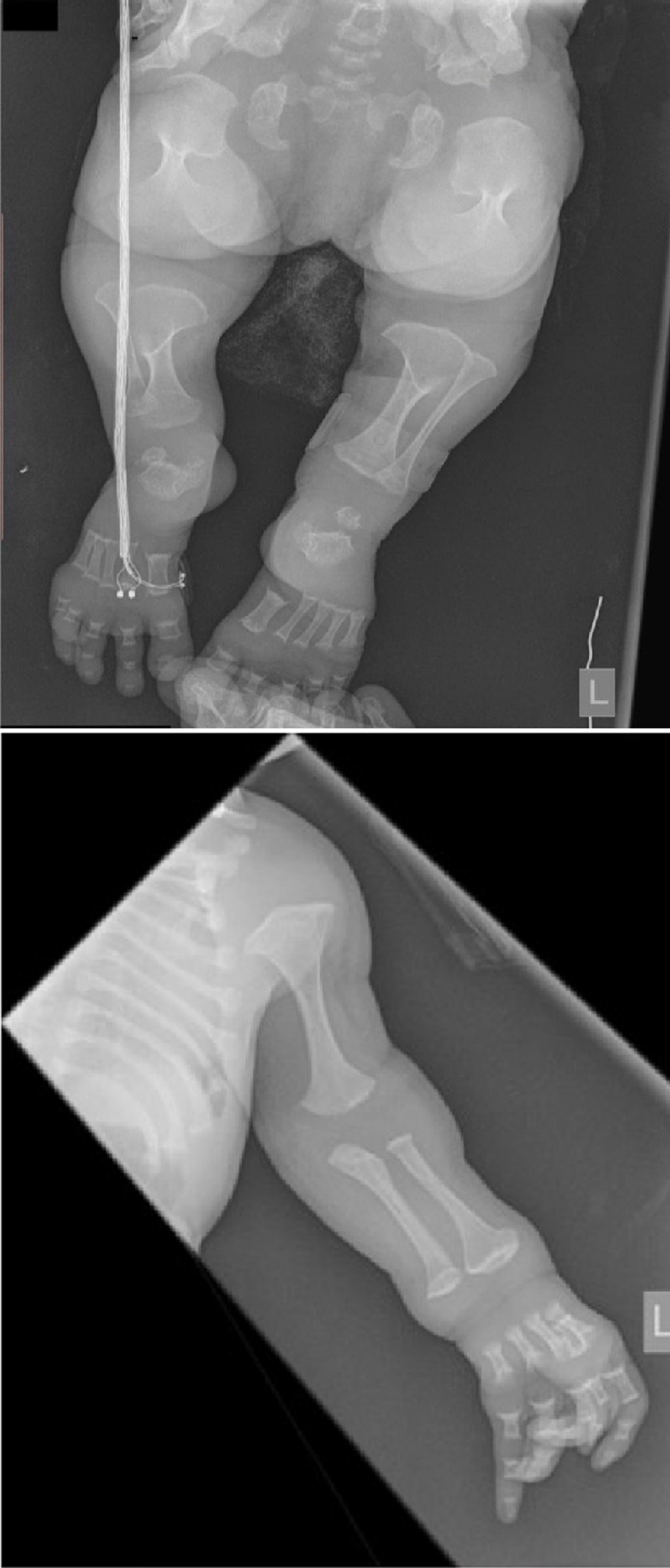 Click for large image | Figure 1. Lower and upper limbs, extremely short long bones, resembling dumbbells, metaphyseal flaring, and hypoplasia of ilium shafts. |
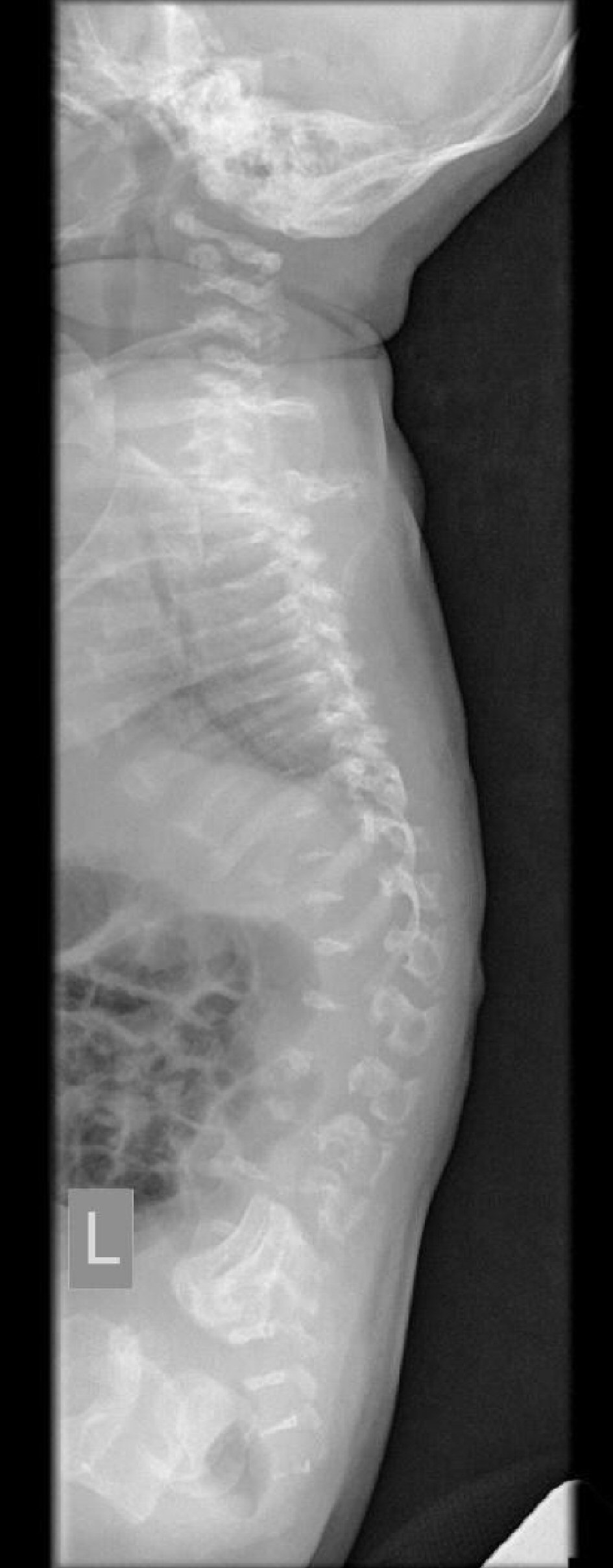 Click for large image | Figure 2. Spine: thin, rounded vertebral bodies, significant narrowing in the dorsal part (platyspondylia), and remaining coccyx. |
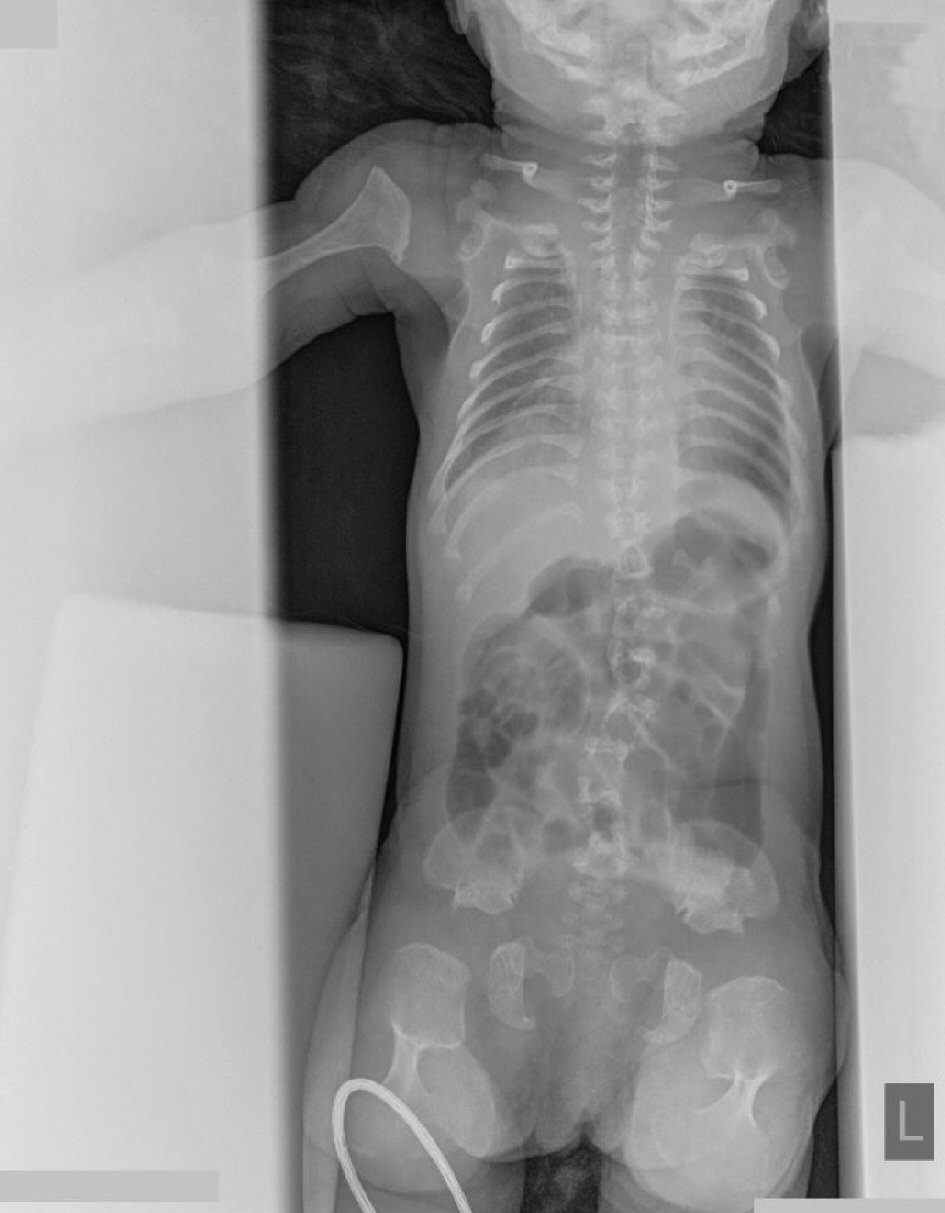 Click for large image | Figure 3. Chest: narrow and elongated chest with short ribs. |
During the stay at the clinic the child had multiple consultations: orthopedic, rehabilitative and radiologic with a hypothesis of spondylometaphyseal dysplasia. Moreover, cardiological, neurological and genetic consultations were held. During the genetic consultation anthropometric examination and analysis of the family history as well as of the available medical records took place. It was determined that the disease occurred in the family for the first time so the inheritance is unknown. Antley-Bixler syndrome (ABS) was suspected and the analysis of the performed X-rays and CT of the skull for craniostenosis of lambdoid and coronal suture was proposed. In the tested genetic material no gene TRPV4 mutation was discovered. The results of laboratory tests assessing calcium-phosphate metabolism demonstrated a significantly lowered vitamin D3 metabolite level in liver and an increased vitamin D3 metabolite level in kidneys which is presented in Table 1. Additionally urine markers were included in Table 2. The child was given one drop of vitamin D3, approximately 500 IU, but after the analysis of the results the dose was increased to three drops, approximately 1,500 IU.
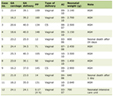 Click to view | Table 1. Calcium-Phosphate Metabolism Assessment in Blood Serum |
Click to view | Table 2. Urine Markers (sample II) |
Following the admission to the ICU the child was in a critical condition, with efficient circulation was intubated, ventilated mechanically (SIMV), quite conscious. The radiological examination revealed atelectatic/infectious lesions; analgosedation was used and then antibiotic treatment was modified based on antibiogram; parenteral feeding was withdrawn and because of the features of anemia, hematopoietic treatment was initiated. Effort of extubated was taken and non-invasive ventilation was used along with caffeine supply. On day 11 of the treatment, due to repeat features of respiratory failure, the girl was intubated again and SIMV was started. On day 15 of the treatment, as there were no signs of infection, antibiotic treatment was stopped and nuclear magnetic resonance (NMR) of the spine was performed that revealed similar lesions concerning the cervical and thoracic spine. Moreover, angular kyphosis of the spine on the thoracic and lumbar border was stated. In the lumbar spine, it was found that the narrowing of the spinal canal at the level of epiphyses of the arches (like in the thoracic segment) up to 4 mm transversely, and 8 mm in AP measurement. The spinal canal modelled on the dorsal side at the level of C1-C2 narrowing in the cervical spine, and the spinal cord with no signs of compression, with normal signal, without focal lesions. In the thoracic segment and on the thoracic and lumbar border, modelled cord especially in the transverse section was found; and low-lying medullary cone is visible at the level of L3. During the hospitalisation the patient had neurosurgical consultation with no recommendations for the surgical procedure. On day 19 of the treatment, antibiotic treatment was implemented again due to clinical features of infection, and was continued for the following 10 days. A pediatric radiology specialist was asked to consult bone X-ray finding, with the following report obtained after the analysis of the collected material: short ribs with broadened rib endings, thin, rounded vertebral bodies, significant narrowing in the dorsal part, hypoplasia of ilium shafts and significant shortening of tabular bones with broadened “mushroom-like” endings. The radiologist made a clear diagnosis of lethal type of metatropic dysplasia also defined as hypochondrogenesis with autosomal recessive inheritance. It is a disorder belonging to the group of lethal dysplasias with platyspondylia.
The 2-month infant in a general fair condition, conscious, intubated, with SIMV of average parameters, with efficient circulation, with gastrostomy tube used for feeding (Table 3), with maintained orthopedic collar was discharged for further treatment at the place of residence, at the Department of Intensive Treatment of the Hospital Polanki in Gdansk. An attempt to contact child’s parents was unsuccessful.
Click to view | Table 3. Anthropometric Measurements |
| Discussion | ▴Top |
The paper presents a rare case of skeletal system defect, metatropic dysplasia; and it is the first case of this disease in our clinical practice. It seems to be important that making a diagnosis based on the clinical picture, phenotypical features and findings of additional examinations, including X-ray, CT and magnetic resonance imaging (MRI) is difficult; and it requires a cooperation between the experts of many medicine specialities, like in the case of the described infant.
The differential diagnostics of metatropic dysplasia should consider ABS, spondylometaphyseal dysplasia or thanatophoric dysplasia (TD).
ABS is a rare type of craniosynostosis, characterised by typical clinical features including: underdeveloped midfacial regions with flat nose, trapezoid face, prominent frontal bossing, proptosis, underdeveloped ears, defects of the musculoskeletal system, such as radiohumeral or radioulnar synostosis, multiple contractures of joints and urinary and genital deficits. ABS is a heterogeneous disorder, so far two types of this syndrome have been described: type 1 involves mutations in FGFR2 (10q26) gene without steroidogenesis disorders, type 2 involves mutations in cytochrome P450 oxidoreductase (POR) coding gene that plays a direct role in steroidogenesis. Type 2 ABS is an autosomal recessive disorder and is associated with abnormal genitalia in both sexes due to impaired steroidogenesis. Occasionally, a diverse range of cardiac, renal, gastrointestinal and vertebral malformations may occur [4, 5]. Our infant has not developed multiple craniofacial malformations, as those described for ABS, nor urogenital defects, no FGFR2 and cytochrome P450 mutation testing was performed which excluded this syndrome as a diagnosis.
TD is a type of neonate dwarfism which is extremely lethal in the perinatal period. It is characterised by short limbs, narrow bell-shaped thorax, macrocephaly with prominent forehead and flattened vertebral bodies. These defects are the result of autosomal dominant mutations in fibroblast growth factor receptor 3 (FGFR3) gene. TD may be divided into two types: type I characterized by micromelia with flattened femurs and by the presence of cloverleaf skull deformity, and type II characterized by micromelia with straight femurs and a moderate to severe cloverleaf skull deformity. The common feature of both types is the presence of narrow thorax which causes respiratory failure soon after birth [6, 7]. Our infant had no flattened femurs described for type I TD, nor flat femurs present in type II TD, no cloverleaf skull deformity was stated, so this syndrome was excluded as a diagnosis.
Spondylometaphyseal dysplasia, Kozlowski type, is a relatively common autosomal dominant disease in a heterogeneous group of approximately 30 different disorders with vertebral and tabular bone metaphyses abnormalities. It is one of the best clinically defined spondylometaphyseal dysplasias with molecular basis explained. TRPV4 mutations identified in patients with this disorder affect calcium-permeable ion channels. These mutations of bone dysplasias are characterised by dwarfism, kyphoscoliosis, distortion and bowing of the extremities, and contractures of the large joints. This disease is characterised by a combination of decreased bone density, bowing of the long bones, platyspondyly and irregularities of endochondral ossification with areas of calcification and streaking in epiphyses, metaphyses and apophyses [8, 9]. The observed neonate had no TRPV4 gene mutation, long bones had “mushroom-like” epiphyses, however without bowing, moreover no streaking in the epiphyses was described, therefore this syndrome was also excluded as a diagnosis.
In the described case the respiration was maintained using SIMV, which enabled to discharge the child to continue treatment at the hospital at her place of residence. Also monitoring of biochemical and calcium-phosphorus metabolism assessing parameters is important to supply adequate doses of vitamin D3 or possibly calcium. A significant issue that we wish to emphasize is the possibility to perform corrective surgeries to stop malformation progression; however it depends on the moment of such an intervention as well as on the careful selection of patients, especially the assessment of pulmonary function. It seems that in the case presented in the paper there will not be such an opportunity, and bone deformities will progress, and probably increase respiratory disorders. The understanding of the clinical spectrum of metatropic dysplasia, and adequate intervention at an early disease stage, mainly through rehabilitation and normal nutrition, may improve patients’ functionality, enabling at the same time their independent social life in difficult cases [1].
Conclusions
Due to a non-specific clinical picture, the diagnostics of metatropic dysplasia is very complicated, sometimes long-lasting; and the diagnosis based on the clinical, phenotypic traits and findings of additional examinations, including X-rays, CT, MRI, requires a close cooperation between doctors of many specialities. Patients require the monitoring of biochemical parameters and vital signs, due to respiratory disorders and progression of bone deformities. An important element is the selection of adequate treatment, including surgical or neurosurgical treatment, to improve the functioning and independent living in the society, as well as providing children’s parents with suitable psychological care.
Acknowledgments
We would like to thank the parents of the girl described for allowing us to share her details and thank PhD MD Tadeuszowi Bieganskiemu for radiological consultation and laboratory in Warsaw for genetic testing.
Financial Disclosure
This work was supported by Medical University of Lodz, Poland (503/1-090-02/503-11-001).
Conflict of Interest
None to declare.
Informed Consent
Not applicable.
Author Contributions
Study design: Izabela Michalus, Agnieszka Byrwa. Data collection: Izabela Michalus, Paulina Adamiecka. Data interpretation: Elzbieta Jakubowska-Pietkiewicz, Izabela Michalus, and Agnieszka Byrwa. Manuscript preparation: Agnieszka Byrwa. Literature search: Izabela Michalus, Agnieszka Byrwa. Funds collection: Elzbieta Jakubowska-Pietkiewicz.
| References | ▴Top |
- Song HR, Sinha S, Song SH, Suh SW. A case of metatropic dysplasia: operative treatment of severe kyphoscoliosis and limb deformities. Oman Med J. 2013;28(6):445-447.
doi pubmed - Orphanet - website, French National Institute for Health and Medical Research.
- Graversen L, Haagerup A, Andersen BN, Petersen KK, Gjorup V, Gudmundsdottir G, Vogel I, et al. Novel TRPV4 variant causes a severe form of metatropic dysplasia. Clin Case Rep. 2018;6(9):1774-1778.
doi pubmed - Solem RC, Martz M, Weiss E, Reese P, Kawamoto H, Lee JC. Multidisciplinary Treatment of Antley-Bixler Syndrome. Cleft Palate Craniofac J. 2017;54(1):100-108.
doi pubmed - Boia ES, Popoiu MC, Puiu M, Stanciulescu CM, David VL. Antley-Bixler syndrome: surgical management of ambiguous genitalia - a case report. Med Princ Pract. 2014;23(4):384-386.
doi pubmed - Calongos G, Hori M, Ogino M, Sawai H. A Case of Thanatophoric Dysplasia Type I with Fetal Hydrops in the First Trimester. Case Rep Obstet Gynecol. 2016;2016:1821230.
doi pubmed - Tan AP, Priego G. Thanatophoric dysplasia type 1 with tectal plate dysplasia and aqueductal stenosis. Childs Nerv Syst. 2019;35(6):1059-1061.
doi pubmed - Bieganski T, Beighton P, Lukaszewski M, Bik K, Kuszel L, Wasilewska E, Kozlowski K, et al. SMD Kozlowski type caused by p.Arg594His substitution in TRPV4 reveals abnormal ossification and notochordal remnants in discs and vertebrae. Eur J Med Genet. 2017;60(10):509-516.
doi pubmed - Kang SS, Shin SH, Auh CK, Chun J. Human skeletal dysplasia caused by a constitutive activated transient receptor potential vanilloid 4 (TRPV4) cation channel mutation. Exp Mol Med. 2012;44(12):707-722.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.