

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website http://www.jofem.org |
Case Report
Volume 3, Number 1-2, April 2013, pages 29-37
Pitfalls in the Diagnosis of Ectopic Adrenocorticotropic Hormone Syndrome: A Case Report
Ana Coelho Gomesa, g, Sonia do Valea, b, Joao Martin Martinsa, b, Ana Filipa Martinsa, Joao Rebelo de Andradec, Jorge Cruzd, Ulrich Welsche, Emilia Oliveiraf, Isabel do Carmoa
aEndocrinology Diabetes and Metabolism Department, Santa Maria Hospital, Lisbon, Portugal
bEndocrinology Department, Lisbon Medical School, University of Lisbon, Portugal
cSurgery Department, Luz Hospital, Lisbon, Portugal
dCardiothoracic Surgery Department, Santa Maria Hospital, Lisbon, Portugal
eMunich Medical School, Pathology Department, Munich, Germany
fPathology Department, Santa Maria Hospital, Lisbon, Portugal
gCorresponding author: Ana Coelho Gomes, Servico de Endocrinologia Diabetes e Metabolismo, Piso 6, Hospital de Santa Maria e Faculdade de Medicina de Lisboa, Av. Professor Egas Moniz, 1649-035 Lisboa, Portugal
Manuscript accepted for publication January 31, 2013
Short title: Ectopic Cushing’s Syndrome
doi: https://doi.org/10.4021/jem145w
| Abstract | ▴Top |
Ectopic production of adrenocorticotropic hormone by carcinoid tumours is uncommon and covert and occult presentations often hamper its diagnosis. A 27-year-old man was admitted with multiple vertebral bodies collapse related to osteoporosis along with other clinical features consistent with Cushing’s syndrome. Both basal and dynamic laboratory evaluation suggested adrenocorticotropic-dependent Cushing’s syndrome diagnosis, specifically ectopic adrenocorticotropic syndrome. Laboratory testing also showed high serum levels of chromogranin A and urinary 5-hydroxyindoleacetic acid. Pituitary and thoracic-abdominal-pelvic imaging proved to be normal, including the octreoscan. To reduce cortisol levels, and before resorting to adrenalectomy, metyrapone was prescribed but eucortisolism was not achieved. Octreotide therapy reduced plasma adrenocorticotropic hormone and cortisoluria to half although the octreoscan had been negative. As no primitive tumour had been detected, the patient underwent bilateral adrenalectomy. Two years and a half after the diagnosis, an octreoscan detected a lung tumour, which was resected. Pathological analysis revealed an atypical carcinoid tumour and immunohistochemistry was positive for adrenocorticotropic hormone but negative for corticotropin-releasing hormone and chromogranin A. Clinical and biochemical resolution of Cushing’ syndrome followed but chromogranin A levels remained above normal. This case highlights both that, although rare, an exuberant osteoporosis can be the initial presentation of ectopic adrenocorticotropic hormone syndrome and high chromogranin A levels may have other origin than direct tumour production. It also raises the question of prescribing somatostatin agonists in patients whose octreoscan is negative as small tumours may produce false negative results. Moreover, the reduction of hypercortisolism before surgery may improve the surgical outcome and with bilateral adrenalectomy may contribute to the primary tumour growth and detection.
Keywords: Ectopic Cushing’s syndrome; Carcinoid tumor; Osteoporosis; Chromogranin A; Hipokalaemia; Octreotide
| Introduction | ▴Top |
Cushing’s syndrome (CS) may broadly be divided into adrenocorticotropic hormone (ACTH) independent CS and ACTH-dependent CS [1]. 12% to 17% of the CS occurrences are due to the ectopic ACTH syndrome (EAS), which are often tumours of neuroendocrine origin [2]. EAS not only includes tumours that secrete ACTH but also those that produce corticotropin-releasing hormone (CRH), which are extremely rare [2-4].
EAS may be caused by several tumours, mainly those that have their origin in neuroendocrine cells of the respiratory or gastrointestinal tract [2, 5, 6]. The most frequent aetiologies of these tumours are small cell lung carcinoma (SCLC), carcinoid tumours (especially those of the lungs, thymus and gastrointestinal tract), islet cell tumours and pheochromocytoma [2]. Carcinoid tumours alone are responsible for 25% of all EAS [2, 7] and they are by far the most frequent cause of EAS with the classical features and habitus of CS. In most cases, when EAS is caused by malignant tumours, circulating ACTH and cortisol levels are extremely high without classical signs of CS but frequent hypokalaemia. Then the onset of CS is sudden, the duration of symptoms short and the clinical phenotype atypical [1, 4, 7]. Conversely, patients who suffer from neuroendocrine tumours (NETs) namely carcinoid tumours or pheochromocytoma usually exhibit the typical cushingoid habitus and have longer clinical courses because of the slow growth of the tumour [1, 4, 7]. Since non-adrenal tumours may produce large amounts of ACTH and other proopiomelanocortin (POMC) derived peptides, baseline steroid values are usually significantly high and increased skin pigmentation may occur [1, 4, 7].
Besides increased ACTH levels, in EAS due to ectopic ACTH secretion there is no plasma and urinary cortisol suppression in high-dose dexamethasone suppression test and typically no ACTH and cortisol rise in both CRH and metyrapone tests. Furthermore, the pituitary scan magnetic resonance imaging (MRI) is expected to show no changes and the inferior petrosal sinus venous sampling (IPSS) shows no central/peripheral ACTH gradient [1, 4, 7-9]. Therefore, additional imaging procedures have to be carried out in order to identify the ACTH or CRH secreting tumour [1, 4, 7-9].
EAS treatment depends on its cause. If the tumour can be found and the disease remains localised its excision can lead to cure [1]. On the contrary, if the ectopic source of ACTH cannot be found it may be necessary to perform bilateral adrenalectomy followed by a careful observation of the patient (sometimes for several years) as the primary tumour can take time to grow and be localised [1]. Medical therapy may be necessary to partially correct the cortisol excess, hypokalaemic alkalosis, high blood pressure and diabetes mellitus (DM) [1].
We describe a case of an EAS caused by ectopic ACTH secretion from an atypical carcinoid lung tumour. It shows several unusual features that may be of value for other cases: steroidogenesis inhibitors should always be used, even if bilateral adrenalectomy is considered the first treatment option or the primary tumour is already localised and is going to be excised; somatostatin analogues may be a valid alternative even in patients with negative octreoscan; bilateral adrenalectomy contributes to the growth of the primary tumour and its localisation; chromogranin A (CgA) is not specific for NETs and its increase may have other origin than direct tumour production in patients suffering from NETs.
| Case Report | ▴Top |
A 27-year-old Caucasian man was admitted to the hospital because of severe hypokalaemia (1.6 mEq/L) in February 2006.
In November 2005 the patient noticed acne and moon face; in January 2006 the onset of paraesthesias on both hands and feet, polydipsia and polyuria; and in February 2006, two weeks before admission, fatigue and generalized weakness. Two days before admission the patient fell from his standing height and was admitted to the emergency room complaining of generalized weakness and lumbosacral pain. He denied weight changes, psychiatric symptoms or to have taken drugs recently.
On admission, the physical examination showed centripetal distribution of the fat tissue (BMI 28.4 kg/m2), moon face, buffalo hump, facial acne, dispersed small bruises on the trunk, generalized weakness and walking inability. The remaining physical examination was normal, including blood pressure levels.
This clinical setting suggested CS diagnosis. Laboratory testing pointed to an ACTH-dependent CS, suggestive of EAS, with highly increased ACTH and cortisol levels, high delta 4-androstenedione serum levels, severe hypokalaemia, hyperglycaemia, hypocalcaemia, hypophosphataemia and metabolic alkalosis (pH: 7.51; PaCO2: 37 mmHg; PaO2: 76 mmHg; HCO3- 29 mmol/L) (Table 1A, B)
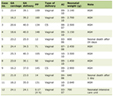 Click to view | Table 1A. Laboratory Testing on Admission |
Click to view | Table 1B. Laboratory Testing on Admission |
The low-dose dexamethasone suppression test (0.5 mg of dexamethasone every 6 hours during 48 hours starting at 8 a.m.) showed no cortisol suppression confirming the CS diagnosis. Although cortisol serum levels were reduced to half at the end of the high-dose dexamethasone suppression test (2 mg of dexamethasone every 6 hours during 48 hours starting at 8 a.m.), they remained extremely high and there was no reduction in the urinary cortisol levels suggesting an EAS. CRH (100 µg IV of CRH and ACTH measurement at 0’, 15’ and 30’) and metyrapone (3g of metyrapone at midnight with ACTH measurement at 8 a.m.) tests showed no ACTH rise (Table 2) and tumour markers determination showed increased levels of CgA, carcinoembryonic antigen (CEA), cancer antigen (CA) 15.3 and 5-hydroxyindoleacetic acid (5-HIAA) (Table 1B), both reinforcing an EAS.
Click to view | Table 2. Low and High-Dose Dexamethasone Suppression Tests; CRH Stimulation Test |
Pituitary MRI was normal. Thus a thorax, abdomen and pelvis MRI and an octreoscan were performed to find out the ectopic tumour. As no pituitary or non-pituitary tumours were detected, an IPSS was carried out. It showed no ACTH production lateralization, no central/peripheral ACTH ratio and no ACTH and cortisol increase after CRH stimulation, reinforcing the hypothesis of an ectopic origin of ACTH (Table 3).
Click to view | Table 3. Inferior Petrosal Sinus Sampling With 1 µg/kg of Endovenous CRH at 0’ |
The Computed Tomography (CT) scan of the dorsolumbar column highlighted the collapse of several vertebral bodies and vertebral sclerosis. In the vertebral column MRI there were non-recent reduction, intra-spongy herniation and degenerative changes of the dorsolumbar vertebral bodies and moderate hypertrophy of the posterior central epidural fat on the dorsal segment, without compression effect. The osteodensitometry showed vertebral osteoporosis with a T-score of -3.3.
Because of severe hypokalaemia, the patient was initially treated with spironolactone (100mg twice a day) and high doses (120 mEq/day) of potassium chloride (KCl). After the EAS diagnosis, metyrapone (500 mg three times a day) was added. In spite of an initial normalization of serum potassium and urinary cortisol, metyrapone dose had to be titrated as its effect was overcome. For the secondary DM, metformin was prescribed and a good glycaemic control was achieved. Because of severe osteoporosis with multiple vertebral fractures, generalized weakness and dorsolumbar pain, treatment with calcium carbonate, alendronate and non-steroid anti-inflammatory drugs (NSAIDs) was started but soon opioids had to be added. The patient also started a proton-pump inhibitor (Omeprazole 20 mg, once a day), physical therapy and a dorsolumbar vest was applied.
During the coming months a progressive deterioration of the clinical condition and worsening of analytical indexes occurred. The patient complained of severe tiredness, inability to walk (a wheelchair was needed), paraesthesias on the lower extremities and bruises for very little traumatisms. Broad violaceous cutaneous striae arose on the abdomen, peri-armpit regions and tights and a severe oedema of the lower extremities extended as far as the tights. ACTH plasma levels, cortisol serum levels and cortisoluria increased and the doses of metyrapone, spironolactone and opioid analgesics had to be titrated. Somatostatin (Sandostatin LAR® 20 mg per month) was added, with a reduction of cortisoluria to half one month later.
But even with the reduction in cortisoluria, no medical control of the CS was achieved, so the patient was proposed to bilateral adrenalectomy, which was performed in May 2007. After the surgery the laboratory tests showed serum hypocortisolism (0.9 µg/dL; reference range - 4.3 - 23 µg/dL) and high levels of plasma ACTH (744 pg/mL; reference range - 0 - 46 pg/mL). As a result, replacement therapy with hydrocortisone was prescribed and later fludrocortisone was added. Calcium carbonate, colecalciferol and alendronate continued being prescribed and only NSAID’s were needed to control pain but they were progressively discontinued.
In September 2007 he underwent a D6-D7-D8 laminectomy with removal of posterior epidural fat. In August 2008 there were no signs of hypercortisolism, DM, oedema or paraesthesias. Besides, there was a reduction of the pain complaints and he was able to walk with the vest. There was a generalised cutaneous hyperpigmentation and increased CEA (9.2 ng/mL - reference range: < 5.0 ng/mL) and CgA (204 µg/L - reference range: < 100µg/L) levels, although they were lower than those at initial presentation (CEA: 17.6 ng/mL; CgA: 1,473 µg/L).
In August 2008, two years and a half after the initial symptoms, he performed another octreoscan which showed a single nodular lesion in the inferior third of the left hemithorax medial surface (Fig. 1) and a thoracic MRI which revealed a 1.8 cm of diameter, nodular lesion in the inferior left lobe of the pulmonary parenchyma (Fig. 2). The nodule excision was performed in September 2008 with no intercurrences. Isolation and section of the lobar bronchus was also carried out. The extemporaneous exam showed a carcinoid tumour and the pathological analysis diagnosed a 2.5 × 2 cm atypical carcinoid tumour with intense and diffuse immunoreactivity to cytokeratines 7, 18 and CD56, positivity to ACTH and synaptofisin and negativity to CRH, CgA and thyroid transcription factor-1 (TTF-1) (Fig. 3). There was minimal necrosis, with the tumour extending to the surgical margins but no angioinvasion. There was tumour tissue in one of the hilar fragments of the left lung inferior lobe and in one of the three lymph nodes that were examined.
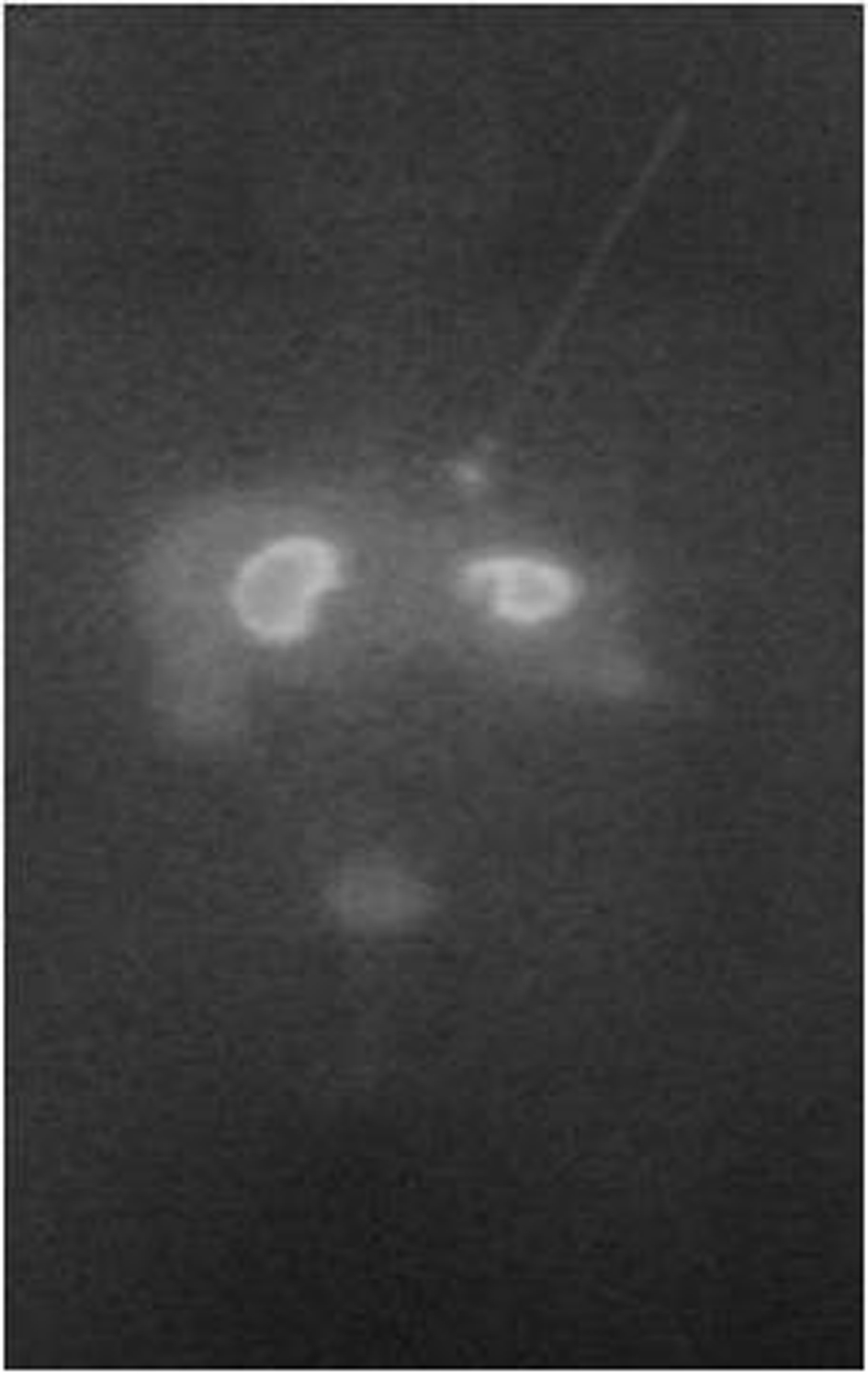 Click for large image | Figure 1. Octreoscan showing a single nodular lesion in the inferior third of the left hemithorax medial surface. |
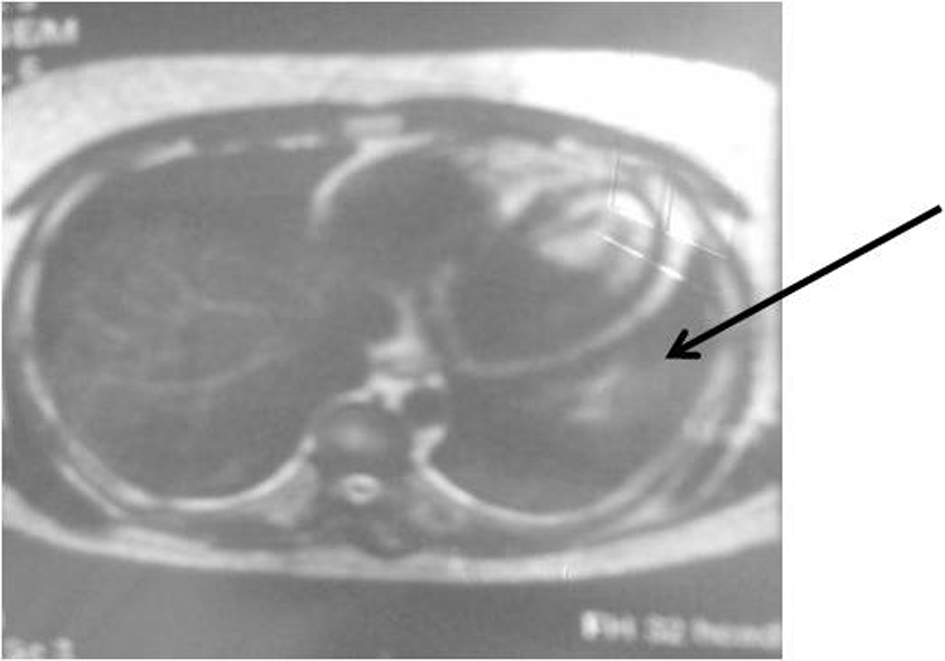 Click for large image | Figure 2. Thoracic MRI showing a 1.8 cm of diameter, nodular lesion in the inferior left lobe of the pulmonary parenchyma. |
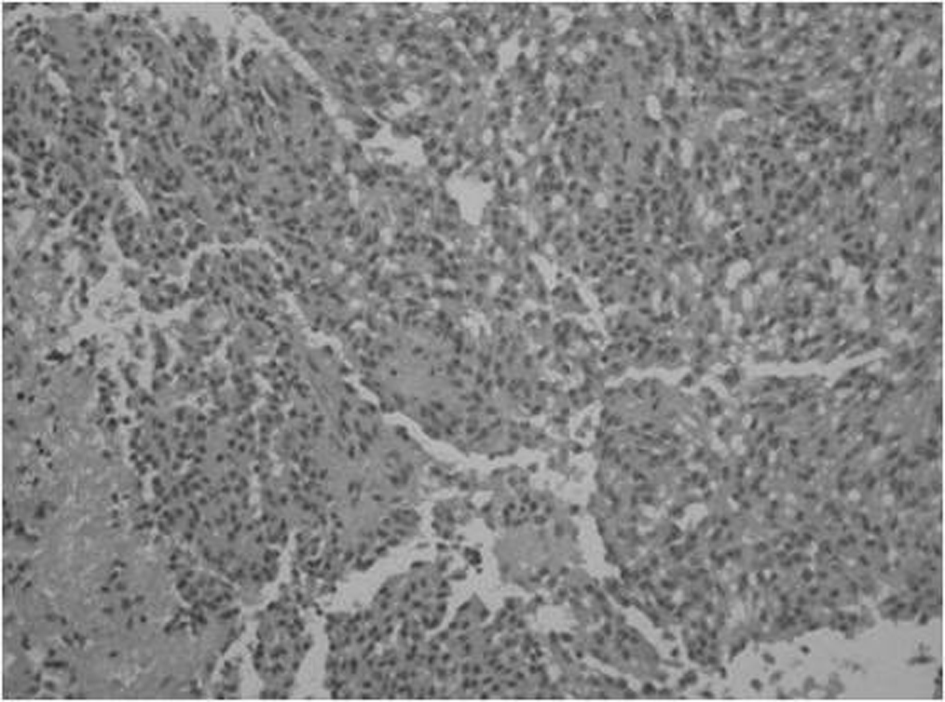 Click for large image | Figure 3. Atypical carcinoid lung tumour × 200. |
Two months later the patient had no complaints, the skin was less pigmented and the ACTH plasma levels had normalised (27 pg/mL - reference range: 0 - 46 pg/mL). However, CA 15.3 was still slightly high (34 U/mL - reference range: < 32 U/mL) and the CgA levels had increased (816 µg/L - reference range: < 100 µg/L). Seven months after the lung surgery, an octreoscan, a bronchofibroscopy and a thorax and abdomen CT scan were carried out but no tumour evidence was found. Although the osteodensitometry showed lumbar column osteopenia (T-score -1.6), there had been an improvement regarding to the one performed by the time the CS was diagnosed (T-score -3.3).
In October 2011, about five years and a half after the EAS diagnosis and three years and a half after the primitive tumour resection, the patient was treated with hydrocortisone, fludrocortisone, calcium carbonate and colecalciferol. He continued asymptomatic, with normal ACTH and cortisol levels, with CA 15.3 and CgA within the reference range and he repeated an octreoscan which was negative. As a precaution, before collecting the blood samples for analysis, the patient stopped the proton pump inhibitor therapy as it may be a cause of CgA false positives.
| Discussion | ▴Top |
This case describes a patient with an EAS secondary to an atypical carcinoid lung tumour, which corresponds roughly to 10% of all lung carcinoid tumours [6]. Lung carcinoid tumours are non-smoking associated rare tumours, accounting for 1-5% of all pulmonary neoplasms and 12-15% of carcinoid tumours overall [2, 10, 11]. They are more frequent in Caucasian males, the male-to-female ratio is 2:1, and affect individuals in a wide age range with a peak incidence in the fifth decade [5, 6]. They have their origin in the bronchial mucosa, in the neurosecretory cells - Kulchistky cells - and are classified as low grade malignant neoplasms because they can potentially cause both local invasion and local recurrence and the occurrence of occasional metastases in extrathoracic sites [5, 6]. Lung carcinoids are ranked typical and atypical, according to the histopathological criteria of the World Health Organization (WHO) [10] and, at present, their staging is named the same way as the lung bronchogenic carcinoma [6, 12]. The incidence of CS in lung carcinoid tumours is about 1% [10]. Conversely, the incidence of lung carcinoid tumours as the cause of CS remains variable in the literature [10].
In February 2006, when admitted to the hospital, our patient had features typical of EAS caused by NETs.
The long-standing disease with the typical cushingoid habitus and associated osteoporosis was one of these features. Indeed, in a retrospective study carried out by Libuse Tauchmanova, Rosario Pivonello, Carolina Di Somma et al, patients with ectopic and pituitary ACTH hypersecretion had more severe cortisol excess than those ones with adrenal adenomas or carcinomas and bone fractures were described as a presenting symptom of endogenous cortisol excess [13, 14]: 4 patients out of a total of 104 (4%) presented low-energy fractures at the time of diagnosis and 38 (37%) medium or high energy fractures [14]. Besides, the severe disease with very high ACTH (higher than 200 pg/mL) and cortisol levels, high serum levels of delta-4-androstenedione, hypokalaemia and metabolic alkalosis and the non-suppression of cortisol in low and high-dose dexamethasone suppression tests and failure of CRH and metyrapone tests to increase ACTH and cortisol levels also suggested an EAS caused by a NET. This diagnosis was reinforced by the CgA levels increase and by the absence of image in the pituitary MRI and no central/peripheral ACTH gradient in the IPSS. Actually CgA is a neuroendocrine marker that may reflect the tumour size, activity and malignancy [1, 15] and that may be 1,000 times higher than the normal reference range in NETs [15]. IPSS, with a sensitivity of more than 95% and very rare false-positive results [4], has proved to be a highly sensitive test regarding the distinction between pituitary and non-pituitary sources of ACTH excess [1, 4, 7, 16], as opposed to pituitary MRI in which false negatives due to Cushing’s disease (CD) microadenomas are common [16, 17].
So, once the EAS diagnosis had been made, its aethiology had to be determined and the primary tumour localised.
Increased levels of urinary HIAA suggested a carcinoid tumour, as it was posteriorly confirmed. Indeed, carcinoid tumours are a common cause of EAS and up to one quarter of lung carcinoids are associated with increased levels of urinary HIAA [17]. Also, the initial negative thorax, abdomen and pelvis MRI and octreoscan pointed to carcinoid tumour diagnosis. In fact, this is a common situation in these tumours as they remain radiologically undetectable for long periods after the initial presentation [18]. The absence of tumour detection in these exams also pointed to the lack of sensibility of these methods in the detection of small carcinoid tumours. Actually, their sensibility is only 49% for the detection of slow growth ectopic ACTH-secreting tumours [18].
High CAE and CA 15.3 levels at presentation pointed to an EAS, but they didn’t help finding its aethiology. In fact, and although CAE and CA 15.3 might be increased in lung cancer, they are non-specific tumour markers and may be raised in many other cancers and other non-cancerous conditions, all excluded in our patient [19].
In conclusion, we would like to point out some unusual features that might be useful for other cases.
The importance of reducing hypercortisolism with medical adrenalectomy before surgery (bilateral adrenalectomy, primary tumour excision or other surgeries). Actually it reduces the surgical risk as the reduction in serum cortisol levels ameliorates the surgical prognosis [20]. Thus, we suggest that all patients with hypercortisolism should be treated with steroidogenesis inhibitors even if bilateral adrenalectomy is considered the first option or the primary tumour is already localised and is going to be excised.
The presence of somatostatin receptors in many ectopic ACTH-secreting tumours is the basis for the use of somatostatin analogues to treat this condition and the uptake of radiolabelled somatostatin has been found to be predictive of biochemical response to somatostatin analogues [18]. In this case a marked and early response to somatostatin analogues was noted despite a negative octreoscan. Although we did not further pursue this therapy and instead choose to perform bilateral adrenalectomy it is worth noting that medical therapy with somatostatin analogues may be a valid alternative despite a negative octreoscan.
After bilateral adrenalectomy increased levels of ACTH were found and the primary tumour was localised about one year later. This is reminiscent of a Nelson-like syndrome, whereby bilateral adrenalectomy, reducing the negative feedback of cortisol on the pituitary leads to an increase activity of the pituitary tumour/corticotroph cells or primary ectopic syndrome. In this regard, the previous note may be particularly relevant since maintained medical therapy with somatostatin analogues may have a similar effect and may lead to the identification of the primary tumour, thus avoiding unnecessary bilateral adrenalectomy. We suggest this could become a rational approach to non-localised EAS.
A fourth worth noting point regards the misleading value of CgA. This is a somehow specific marker of neuroendocrine cells/tumours mostly produced but chromaffin cells of the adrenal medulla, but also synthesized in anterior pituitary gland and pancreatic β cells, what explains its rise in DM. Besides DM, CgA levels are increased in renal and heart failures, chronic atrophic gastritis, osmotic diarrhea and therapy with proton-pump inhibitors [21]. Glucocorticoids also raise CgA levels [22, 23] and CgA secretion from the adrenal medulla and anterior pituitary gland into the blood stream is increased during stress [15]. In fact, and although produced in the cortex, glucocorticoids reach the adrenal medulla through the corticomedullary portal system and, at high levels, can promote the upregulation of phenylethanolamine N-methyltransferase (PNMT), increasing epinephrine synthesis and secretion and therefore raising CgA [1]. Although the high levels found in this patient were in apparent agreement with a carcinoid tumour, later data clearly show the tumour was not the origin of the increased CgA levels - no immunoreactivity for CgA in the tumour itself and persistent high levels after tumour excision. This may induce unnecessary anxiety and unneeded further exploration. In fact, increased CgA in this patient probably depends as noted on the glucocorticoid effects on CgA secretion, DM and proton-pump inhibitors use, as, and excluding the initial and final CgA measurements (February 2006 and October 2011 respectively), the patient was taking proton-pump inhibitors.
Acknowledgments
The authors thank Antonio Alves, medical doctor from the Pathology Department of Santa Maria Hospital for providing us the photographs of the lung sections.The authors thank Mrs. Maria de Lurdes Gomes for the English language review.
| References | ▴Top |
- Stewart PM. The adrenal cortex. In: Kronenberg HM, Melmed S, Polonsky KS, Larsen PR (eds). Williams Textbook of Endocrinology. 11th edn. Philadelphia: Saunders Elsevier, 2008: 464-477.
pubmed - Isidori AM, Lenzi A. Ectopic ACTH syndrome. Arq Bras Endocrinol Metabol. 2007;51(8):1217-1225.
doi - Shahani S, Nudelman RJ, Nalini R, Kim HS, Samson SL. Ectopic corticotropin-releasing hormone (CRH) syndrome from metastatic small cell carcinoma: a case report and review of the literature. Diagn Pathol. 2010;5:56.
doi pubmed - Jameson JL. Disorders of the adrenal cortex. In: Braunwald E, Fauci AS, Hauser SL, Kasper DL, Longo DL, Jameson JL, Loscalzo J. (eds). Harrison's Principles of Internal edicine. 17th edn. USA: McGraw-Hill, 2008: 2254-2259.
- Khan AN, Ghanem SA, Irion KL, MacDonald S, Allen CM. Lung, carcinoid. Emedicine 2010. (http://emedicine.medscape.com/article/357921-overview).
- Mancini MC. Carcinoid lung tumors. Emedicine 2010. (http://emedicine.medscape.com/article/426400-overview).
- Morris DG, Grossman AP, Nieman LK. Cushing's syndrome. In: Jameson JL, De Groot LJ, Grossman AP, Kretser D, Marshall JC, Melmed S, Potts JT et al. Endocrinology Adult and Pediatric. 6th edn. Philadelphia: Saunders Elsevier, 2010: 282-283.
- McPhee SJ. Disorders of the Adrenal Cortex. In: McPhee SJ, Ganong WF (eds). Pathophysiology of Disease: an Introduction to Clinical Medicine. 5th edn. USA: McGraw-Hill, 2006: 598-607.
- Adler GK. Cushing's syndrome. Emedicine 2010. (http://emedicine.medscape.com/article/117365-overview).
- de Matos LL, Trufelli DC, das Neves-Pereira JC, Danel C, Riquet M. Cushing's syndrome secondary to bronchopulmonary carcinoid tumor: report of two cases and literature review. Lung Cancer. 2006;53(3):381-386.
doi pubmed - Atoui R, Almarzooqi S, Saleh W, Marcovitz S, Mulder D. Bronchopulmonary carcinoid tumor associated with Cushing syndrome. Ann Thorac Surg. 2008;86(5):1688-1690.
doi pubmed - Jameson J.L. Disorders of the adrenal cortex. In: Braunwald E, Fauci AS, Hauser SL, Kasper DL, Longo DL, Jameson JL, Loscalzo J. (eds). Harrison's Principles of Internal Medicine. 17th edn. USA: McGraw-Hill, 2008: 555.
- Tauchmanova L, Pivonello R, Di Somma C, Rossi R, De Martino MC, Camera L, Klain M, et al. Bone demineralization and vertebral fractures in endogenous cortisol excess: role of disease etiology and gonadal status. J Clin Endocrinol Metab. 2006;91(5):1779-1784.
doi pubmed - Vestergaard P, Lindholm J, Jorgensen JO, Hagen C, Hoeck HC, Laurberg P, Rejnmark L, et al. Increased risk of osteoporotic fractures in patients with Cushing's syndrome. Eur J Endocrinol. 2002;146(1):51-56.
doi pubmed - Taupenot L, Harper KL, O'Connor DT. The chromogranin-secretogranin family. N Engl J Med. 2003;348(12):1134-1149.
doi pubmed - Kaskarelis IS, Tsatalou EG, Benakis SV, Malagari K, Komninos I, Vassiliadi D, Tsagarakis S, et al. Bilateral inferior petrosal sinuses sampling in the routine investigation of Cushing's syndrome: a comparison with MRI. AJR Am J Roentgenol. 2006;187(2):562-570.
doi pubmed - Isidori AM, Kaltsas GA, Mohammed S, Morris DG, Jenkins P, Chew SL, Monson JP, et al. Discriminatory value of the low-dose dexamethasone suppression test in establishing the diagnosis and differential diagnosis of Cushing's syndrome. J Clin Endocrinol Metab. 2003;88(11):5299-5306.
doi pubmed - McDermott JH, Thabit H, Hickey N, Thompson C, Gaffney E, Young V, Sreenan S. ACTH-secreting bronchial carcinoid: a diagnostic and therapeutic challenge. Ir J Med Sci. 2008;177(3):269-272.
doi pubmed - Almeida JRC, Pedrosa NL, Leite JB, Fleming TRP, Carvalho VH, Cardoso AAA. Marcadores Tumorais: Revisao de Literatura. Revista Brasileira de Cancerologia 2007; 53(3):305-316.
- Mancini T, Porcelli T, Giustina A. Treatment of Cushing disease: overview and recent findings. Ther Clin Risk Manag. 2010;6:505-516.
- Biausque F, Jaboureck O, Devos P, D'Herbomez M, Hainaut P, Carre A, Mounier-Vehier C. [Clinical significant of serum chromogranin A levels for diagnosing pheochromocytoma in hypertensive patients]. Arch Mal Coeur Vaiss. 2003;96(7-8):780-783.
pubmed - Rozansky DJ, Wu H, Tang K, Parmer RJ, O'Connor DT. Glucocorticoid activation of chromogranin A gene expression. Identification and characterization of a novel glucocorticoid response element. J Clin Invest. 1994;94(6):2357-2368.
doi pubmed - Hendy GN, Bevan S, Mattei MG, Mouland AJ. Chromogranin A. Clin Invest Med. 1995;18(1):47-65.
pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.