

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website http://www.jofem.org |
Case Report
Volume 3, Number 3, June 2013, pages 73-77
Growth Hormone and Insulin Growth Factor-I insensitivity: Case Report of a Patient With Severe Short Stature
Florbela Ferreiraa, c, Sara Pintob, Carla Pereirab, Brigida Robalob, Leonor Botob, Maria de Lurdes Sampaiob
aEndocrinology, Diabetes and Metabolism Department, Santa Maria Hospital, Av. Professor Egas Moniz, 1649-035 Lisbon, Portugal
bPaediatric Endocrine Unit, Department of Paediatrics, Santa Maria Hospital, Av. Professor Egas Moniz, 1649-035 Lisbon, Portugal
cCorresponding author: Florbela Ferreira, Endocrinology, Diabetes and Metabolism Department, Santa Maria Hospital, Av. Professor Egas Moniz, 1649-035 Lisbon, Portugal
Manuscript accepted for publication June 14, 2013
Short title: Growth Hormone and Insulin Growth Factor-I Insensitivity
doi: https://doi.org/10.4021/jem177w
| Abstract | ▴Top |
Normal GH secretion and functional integrity of the growth hormone (GH) and insulin growth factor (IGF)-I axis are essential for linear growth. Primary GH deficiency causes short stature and, when treated with recombinant human (rh) GH, normal adult height is generally achieved. Cases of GH insensitivity are treated with rhIGF-I, with more modest increases in height. Several genetic defects have been identified in the GH-IGF-I axis. We present the case of a 5-year-old boy with severe short stature (-4.4SDS) and evidence of primary GH deficiency. Treatment with rhGH was ineffective. Latter, rhIGF-I and androgens were sequentially introduced. Growth velocity peaked during treatment with exogenous androgens, but height SDS was persistently between -4.6 and -5.1. At age 17, his height was 140.3 cm (-4.5SDS). Genetic analysis for GH-IGF axis mutations was negative. We believe this patient presents a yet unrecognised mutation affecting multiple sites of the GH-IGF axis. To our knowledge, there are no previously reported cases with this pattern of growth and irresponsiveness to therapy.
Keywords: Severe short stature; Growth hormone deficiency; Insulin growth factor-I insensitivity
| Introduction | ▴Top |
Normal growth hormone (GH) secretion and the functional integrity of the insulin-like growth factor (IGF) system are essential for normal linear growth [1]. GH actions are mediated by the GH receptor, the intracellular signal transduction system and a combination of components of the IGF system, including IGF-I, IGF-binding protein and the IGF-I receptor [1, 2].
The GH-IGF-I axis should be assessed in conditions of severe short stature (< -3 SD), severe growth deceleration (height velocity < -2 SD) or less severe short stature (height between -2 and -3 SD) combined with growth deceleration (height velocity < -1 SD) [3]. Primary GH deficiency (GHD) results in normal birth size but, when untreated, adult final height is severely compromised [4]. A peak GH concentration of less than 10 ng/mL following stimulation by one or more secretagogues and assayed by RIA has traditionally been used to support the diagnosis of GHD [5].
Treatment with exogenous rhGH began in 1985 and has become a well-accepted therapeutic option for children with growth failure caused by GHD, as well as many other conditions [6]. It has been demonstrated to increase both short-term growth and adult height [7].
GH insensitivity (GHI) should be suspected in patients with growth failure and low IGF-I and IGFBP3 levels, but normal or increased circulating levels of GH and, more importantly, lack of growth response to rhGH.
GHI comprises a range of disorders resulting from deficiency of either production or peripheral action of IGF-I on linear growth. While IGF-I deficiency (IGFD) secondary to low GH concentration is typically highly responsive to rhGH therapy (with the rare exception of patients developing anti-GH antibodies), patients with severe primary IGFD generally show no or marginal responses to rhGH, even at pharmacological doses [8].
The only therapy available to date for treatment of severe GHI is recombinant human IGF-I (rhIGF-I), which is available since 1986 but approved by the FDA in 2005, by the EMEA in 2007 and since 2010 in Portugal [9]. Patients usually respond to treatment with IGF-I, but the growth response is substantially less than that of a GH-deficient patient treated with rhGH. This lower effect in growth is likely due to the fact that rhIGF-I fails to replicate the physiological IGF-I distribution and restore the direct effect of GH on growth and metabolism [9].
An American long-term follow-up study of patients treated with rhIGF-1 [10] showed that after the first year of therapy growth velocity greatly decelerated. The authors concluded that patients may achieve a significantly greater adult height than expected in the absence of therapy, but it was unlikely for them to achieve regular adult height, unlike what happens with the treatment with rhGH [11].
Since 1966 more than 250 patients with genetic GHI have been identified worldwide. Most cases have autosomal recessive inheritance and result from homozygous or compound heterozygous mutations [1]. Still, little is known about this field. There are many components of the GH-IGF-I signalling cascades that remain poorly understood and are legitimate candidates for harbouring significant mutations [2].
| Case Report | ▴Top |
A 5.4-year-old male patient was referred to the Paediatric Endocrinology Unit because of severe short stature (height 89.5 cm, < 3rd centile, -4.4 SDS). His weight was proportionately low (11.5 kg, -5.1 SDS).
He was born after 40 weeks of an uneventful pregnancy. His birth weight was 3.13 kg (-0.7 SDS), length 47 cm (-1.1 SDS), and head circumference 33.5 cm (-0.8 SDS). He was diagnosed with Ivemark syndrome in the first day of life, presenting asplenia, right sided isomerism, and severe cardio-circulatory defects, including pulmonary artery atresia, total anomalous pulmonary venous return, malposition of the great vessels and atrioventricular septal defect. He was submitted to multiple cardiothoracic surgeries: BT shunt in the neonatal period, bilateral cavopulmonary shunt at two years of age and total cavopulmonary shunt when he was seven years old. The last post-operative period was complicated by right pleural effusion and systemic infection with E. coli. A transthoracic echocardiogram performed two weeks after surgery revealed good ventricular function, minimal atrioventricular regurgitation and no pericardial effusion. Since then he has been stable in cardiac assessments.
There were no concerns about motor development or cognitive function and there was no other relevant medical history. No dysmorphic features were noted.
Parents are not consanguineous. His father’s height is 175 cm and his mother’s is 151 cm. The patient’s calculated target adult height is 169.4 cm (-1 SDS). He had a 3-year-old sister with normal height for age. Family history was unremarkable.
Investigations initially performed revealed low IGF-I (66.2 µg/L) and slightly elevated TSH (8.74 µUI/mL), with normal fT4 and negative anti-thyroid antibodies. Other endocrine investigations, including ACTH, prolactin and cortisol, were normal. Full blood count, sedimentation rate, albumin, ionogram, calcium, phosphate, renal and hepatic function were normal. Anti-endomysial, anti-gliadin and anti-transglutaminase antibodies were negative. Parasitological examination of faeces was also negative.
The peak value of GH in the L-dopa test was 2.6 µg/L and in the clonidine test was 3.2 µg/L, confirming a GH deficiency.
Chromosome analysis demonstrated a 46 XY karyotype. Radiographic left carpal bone examination revealed a bone age of 3 when the chronological age was 6 years. Cranial MRI showed a small sized sella turcica and adenohypohysis and the expected hyperintensity of the pituitary posterior lobe was lacking. The pituitary stalk was centered and not connected to the pituitary gland and was hyperintense, as was the tuber cinereum, indicating ectopic neurohypophysis.
Figure 1 depicts the basal evaluation and impact of therapy on height, growth velocity, bone age, and endocrine status.
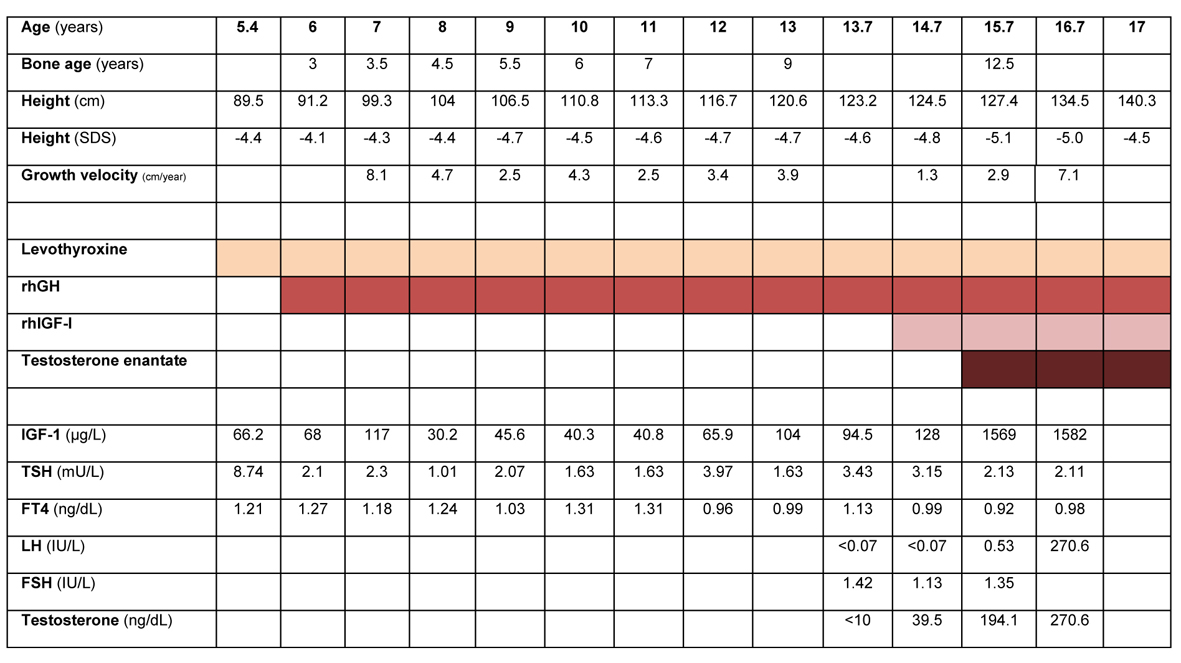 Click for large image | Figure 1. Treatment, laboratory and anthropometric data for age during follow-up of the patient. Reference range (male), IGF-I: 6 - 8 y, 88 - 474; 9 - 11 y, 110 - 565; 12 - 15 y, 202 - 957; 16 - 24 y, 182 - 780 µg/L. Total testosterone: 10 - 11 y, 5 - 50; 12 - 14 y, 10 - 572; 15 - 17 y, 220 - 800 ng/dL. TSH: 0.4 - 5.5 mU/L; FT4: 0.8 - 2.7 ng/dL. LH: 12 - 14.9 y, 0.25 - 4.84; 15 - 18.9 - 0.69 - 7.15 UI/L. FSH: 12 - 14 y, 0.23 - 10.37; 15 - 18 y, 0.81 - 8.18 IU/L. |
Treatment with levothyroxine (25 µg/day) was started after the first endocrine evaluation and TSH was normal after 3 months - Figure 1. Treatment with rhGH (0.025 mg/kg/day) was started when the patient was 6 years old, after approval by the National Committee for the Normalization of Growth Hormone. Adequate administration and compliance to treatment was assured. Dosage was progressively increased to a maximum of 0.035 mg/kg/day. Height velocity increased in the first year of therapy (8.1 cm), but decreased in the following years. Height SDS was persistently below -4. Serial radiographic left carpal bone examination revealed a persistent delay in bone age of 3.5 to 4 years. Levels of IGF-I were consistently below the normal range for age, which prompted consideration of a GHI syndrome, despite initial low levels of GH.
In 2010, when the patient was 14.7 years old, rhIGF-I was added to rhGH therapy. Starting dose was 0.04 mg/kg bid, progressively increased until a maximum dose of 0.12 mg/kg bid. During the two following years, height velocity SDS was between -4.8 and -5.1. Levels of IGF-I were persistently above upper limit of the normal range. During the course of treatment there was no clinical or laboratory evidence of hypoglycemia. There were also no complaints of hypoacusia, snoring, nor clinical evidence of lipohypertrophy, facial dysmorphic features or intracranial hypertension.
Pubertal development was first noticed when the patient was 13.5 years old. Testicular growth was slow and LH and testosterone remained within prepubertal levels for the next 1.5 years. At the age of 15, testicular volume was 6 - 8 mL bilaterally. A LHrH stimulation test was performed, with peak levels of LH of 6.7 UI/L at 30’ and FSH 4.8 UI/L at 90’.
At 15.7 years old, with a bone age of approximately 12.5 years, monthly therapy with intra-muscular testosterone enantate was started. Testosterone levels were successfully kept within the normal range for age. With this combined triple medication, the patient grew 7.1 cm in the following year. In the last follow-up visit, around his 17th birthday, the patient height was 140.3 cm (-4.5 SDS). Genetic testing (GHR, STAT5B, IGF-1, IGFALS, IGF1R, GH1) did not identify genetic abnormalities affecting the GH-IGF axis.
| Discussion | ▴Top |
In the evaluation of severe short stature, it is essential to distinguish GHD from other causes of growth retardation. We could not find evidence of causes of secondary growth deficiency, such as anemia, chronic inflammatory disease, nutritional deficiencies, chronic renal, hepatic or bone disease, celiac disease, intestinal parasitic infection or chromosomopathies [12]. Subclinical hypothyroidism was found but could not explain such a severe growth delay, which persisted after its correction with supplemental levothyroxine. Serial echocardiograms showed normal ventricular function, so the congenital cardiac defect was also excluded as a cause of this severe growth delay.
The MRI findings of a small sized sella turcica and adenohypohysis and ectopic neurohypophysis did not seem to have laboratory and clinical repercussion, as the levels of the remaining anterior pituitary hormones were normal and there was no evidence of diabetes insipida. The link of these findings to the clinical picture of severe growth failure is unclear. Nevertheless, other authors have described an increased frequency of structural malformations of the hypothalamo-pituitary region in patients with GHD, as well as a negative correlation with the levels of GH and IGF-I [13].
Because there is not an established gold standard test, at least two GH stimulation tests are required for the diagnosis of GHD [14]. Diagnosis of GHD is made after a peak GH below 10 µg/L in two tests [15]. In the L-dopa and clonidin stimulation tests, the secretagogues are administered orally after an overnight fast in a dosage adjusted for body weight and there is measurement of serum GH every 30’ until 240’ [16]. Our patient had a peak GH in the L-dopa test after 60’ of 2.6 µg/L and 3.2 µg/L in the clonidine test, which was below the cut-off point and confirmed a state of GHD.
Studies with patients with GHD treated with rhGH have obtained a significant increase in growth velocity, allowing patients to achieve normal adult height [11]. Although this patient had a diagnosis of GHD, there was no increase in linear growth with rhGH, except for the first year of therapy. The patient’s height SDS remained between -4 and -5 in the 7-year period of daily therapy with rhGH, well below the reference range for the general population.
At this point, the possibility of growth hormone insensitivity syndromes was considered. In view of a low GH level, acquired insensitivity by production of anti-GH antibodies was thought. Rare homozygous microdeletions and single base substitutions in the GH1 coding region result in anti-GH antibody formation during GH therapy, neutralizing the growth response and resulting in a state of GHI associated with severe short stature. RhIGF-I becomes the only effective management for the growth failure of these patients [17].
Therapy with rhIGF-I was started as soon as it was available and approved for medical use in our country. Therapy with rhGH was not stopped at this point. With this combined therapy there was still no change in growth velocity or height SDS, despite the increase in dosage. Levels of IGF-I were persistently well above the normal range, raising concerns about possible overdosage, but were probably due to collection of blood during the peak of action of rhIGF-I. There was close monitoring of the expected complications but there was no clinical evidence of major side effects.
Delayed puberty is defined as the start of puberty at a chronological age older than + 2 SD of average maturers. In Europe, in boys, it is diagnosed when testicular growth (testes volume of 4 mL or more) has not started at the age of 14 years. The GnRH/LHrH test is often used as diagnostic tool in the evaluation of delayed puberty [18]. The pubertal growth spurt is decreased in patients with GH deficiency or resistance even with normal testosterone levels. It is speculated that testosterone needs the presence of normal GH secretion to exert its full growth-promoting effect [19]. In boys, stimulation of growth and development of the secondary sex characteristics are usually possible to achieve using gradually increasing doses of testosterone derivates. It should not be started until a skeletal age of 12 years or a chronological age of 14 years is achieved [20].
The results of the LHrH stimulation test in this patient provided evidence of adequate pituitary reserve, with a pubertal pattern of gonadotrophin response: LH peak > 5 U/L, with LH peak greater than FSH peak. However, levels of testosterone were persistently below normal range for age and treatment with testosterone was considered at this point. In the following year, the development of secondary sex traits was accelerated and the patient’s growth velocity almost tripled.
However, at the age of 17, his linear growth is now almost complete and his height is severely decreased (-4.5 SDS), predicting an adult final height significantly below the median for age and gender of the reference population and also below the one predicted from his parents height.
A wide range of genetic defects associated with growth delay have been identified. GHI is now known to be not a single entity but a broad diagnostic category comprising a range of molecular defects in the GH-IGF-I axis. The phenotype of our patient did not match any of the described phenotypes typically associated with genetic mutations of the GH receptor, STAT5B, GH, IGF-I, IGFALS or IGF-I receptor [2, 4]. Genetic analysis was also unable to identify any genetic abnormality in the genes coding for these proteins, rendering this clinical picture difficult to explain.
Conclusions
This patient had a severe growth delay since early childhood, which was attributed to primary GH deficiency. There was no significant impact on growth with treatment with rhGH and later rhIGF-I. At 17 years old, near completion of the linear growth process, the patient’s height is significantly below the predicted adult height and median height for the reference male population. The genetic analysis was negative for the known mutations affecting the GH-IGF1 axis. We admit the possibility of a yet unrecognised mutation affecting multiple sites of the GH-IGF axis.
The field of growth delay and short stature is vast and a myriad of genes are implicated. This case report highlights the fact that our knowledge in this area is still limited and further research is needed to improve our ability to treat patients with severe short stature.
| References | ▴Top |
- Savage MO, Hwa V, David A, Rosenfeld RG, Metherell LA. Genetic Defects in the Growth Hormone-IGF-I Axis Causing Growth Hormone Insensitivity and Impaired Linear Growth. Front Endocrinol (Lausanne). 2011;2:95.
- David A, Hwa V, Metherell LA, Netchine I, Camacho-Hubner C, Clark AJ, Rosenfeld RG, et al. Evidence for a continuum of genetic, phenotypic, and biochemical abnormalities in children with growth hormone insensitivity. Endocr Rev. 2011;32(4):472-497.
doi pubmed - Richmond EJ, Rogol AD. Growth hormone deficiency in children. Pituitary. 2008;11(2):115-120.
doi pubmed - Walenkamp MJ, Wit JM. Genetic disorders in the GH IGF-I axis in mouse and man. Eur J Endocrinol. 2007;157(Suppl 1):S15-26.
doi pubmed - Wilson DM, Frane J. A brief review of the use and utility of growth hormone stimulation testing in the NCGS: do we need to do provocative GH testing? Growth Horm IGF Res. 2005;15 Suppl A:S21-25.
- Richmond E, Rogol AD. Current indications for growth hormone therapy for children and adolescents. Endocr Dev. 2010;18:92-108.
doi pubmed - Lee PA, Germak J, Gut R, Khutoryansky N, Ross J. Identification of factors associated with good response to growth hormone therapy in children with short stature: results from the ANSWER Program(R). Int J Pediatr Endocrinol. 2011;2011:6.
doi pubmed - Rosenfeld RG. IGF-I therapy in growth disorders. Eur J Endocrinol. 2007;157(Suppl 1):S57-60.
doi pubmed - Fintini D, Brufani C, Cappa M. Profile of mecasermin for the long-term treatment of growth failure in children and adolescents with severe primary IGF-1 deficiency. Ther Clin Risk Manag. 2009;5(3):553-559.
- Chernausek SD, Backeljauw PF, Frane J, Kuntze J, Underwood LE. Long-term treatment with recombinant insulin-like growth factor (IGF)-I in children with severe IGF-I deficiency due to growth hormone insensitivity. J Clin Endocrinol Metab. 2007;92(3):902-910.
doi pubmed - Guevara-Aguirre J, Rosenbloom AL, Vasconez O, Martinez V, Gargosky SE, Allen L, Rosenfeld RG. Two-year treatment of growth hormone (GH) receptor deficiency with recombinant insulin-like growth factor I in 22 children: comparison of two dosage levels and to GH-treated GH deficiency. J Clin Endocrinol Metab. 1997;82(2):629-633.
doi pubmed - Jorge AA. [Short stature investigation: clinical, laboratorial and genetic aspects concerning the growth hormone insensitivity (GHI)]. Arq Bras Endocrinol Metabol. 2008;52(6):1056-1065.
doi pubmed - Kucharska A, Boltuc A, Chojnacki J. [The frequency of structural abnormalities in the hypothalamo-pituitary region and their clinical significance in patients with growth hormone deficiency]. Pediatr Endocrinol Diabetes Metab. 2010;16(3):142-147.
- Obara-Moszynska M, Kedzia A, Korman E, Niedziela M. Usefulness of growth hormone (GH) stimulation tests and IGF-I concentration measurement in GH deficiency diagnosis. J Pediatr Endocrinol Metab. 2008;21(6):569-579.
pubmed - Guyda HJ. Growth hormone testing and the short child. Pediatr Res. 2000;48(5):579-580.
doi pubmed - Eren MA, Tabur S, Turan MN, Sanfakiogullari S, Sabuncu T. Comparison of Diagnostic Values of Growth Hormone Stimulation Tests in Adolescents. Turk Jem. 2010; 14: 6-9.
- Riedl S, Frisch H. Effects of growth hormone (GH) and insulin-like growth factor-I therapy in patients with gene defects in the GH axis. J Pediatr Endocrinol Metab. 2006;19(3):229-236.
doi pubmed - Delemarre EM, Felius B, Delemarre-van de Waal HA. Inducing puberty. Eur J Endocrinol. 2008;159 Suppl 1:S9-15.
doi pubmed - Aynsley-Green A, Zachmann M, Prader A. Interrelation of the therapeutic effects of growth hormone and testosterone on growth in hypopituitarism. J Pediatr. 1976;89(6):992-999.
doi - Pozo J, Argente J. Ascertainment and treatment of delayed puberty. Horm Res. 2003;60 Suppl 3:35-48.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.