

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website http://www.jofem.org |
Case Report
Volume 4, Number 1-2, April 2014, pages 39-46
An Unusual Case of a Composite Pheochromocytoma With Neuroblastoma
Oren Steena, d, Judith Fernandob, Jennifer Ramsayc, Ally P.H. Prebtania
aDivision of Endocrinology and Metabolism, McMaster University, 1280 Main Street West, Hamilton, Ontario L8S 4L8, Canada
bDepartment of Internal Medicine, Northern Ontario School of Medicine, 935 Ramsey Lake Road, Sudbury, Ontario P3E 2C6, Canada
cDivision of Anatomical Pathology, McMaster University, 1280 Main Street West, Hamilton, Ontario L8S 4L8, Canada
dCorresponding author: Oren Steen, McMaster University, 237 Barton St E Rm 411, Hamilton, Ontario, Canada
Manuscript accepted for publication March 24, 2014
Short title: Composite Pheochromocytoma
doi: https://doi.org/10.14740/jem211w
| Abstract | ▴Top |
Composite pheochromocytoma (CP) is a rare tumor of the adrenal medulla, consisting of neuroendocrine and neural components. Despite similar neural crest origins, pheochromocytomas and neurogenic tumors are distinct entities. Symptoms may arise from hypersecretion of hormones by either component; however, not all patients present with classic symptoms. Moreover, various medical conditions and substances can confound screening tests and complicate the diagnosis. The patient, a 46-year-old male, was seen in the endocrine clinic regarding a 2-year history of paroxysmal headaches, palpitations and diaphoresis. His medical history was significant for hypothyroidism and substance misuse (nicotine, marijuana and cocaine). Family history was negative for pheochromocytoma, hyperparathyroidism or thyroid malignancies. He was found to be hypertensive in clinic, and two cafe-au-lait spots and bilateral axillary freckles were noted on dermatological examination. Pheochromocytoma was suspected and investigations revealed 24-h urinary catecholamines, metanephrines andvanillylmandelic acid levels that were substantially elevated. He was asked to repeat the urine collection while abstaining from cocaine, given the potential confounding with cocaine-induced elevations of catecholamines and their metabolites; however, the patient did not comply with this. His plasma free metanephrines and chromogranin A were later found to be elevated. Abdominal computed tomography revealed a 5.8 × 5 cm mass in the left adrenal gland, with an attenuation of 40 Hounsfield units. He underwent an uncomplicated laparoscopic left adrenalectomy, after which his blood pressure normalized. Histological features of the tumor revealed findings consistent with a typical pheochromocytoma with neuroblastomatous infiltrates comprising < 5% of the tumor. The mitosis karyorrhexis index was low and there was no ganglioneuromatous infiltrate evident. Medical oncology was consulted in view of the histopathological findings, and no adjuvant therapy was recommended. Unfortunately, he did not fully comply with repeat imaging or biochemical evaluation. Six months postoperatively, the patient remains completely asymptomatic and normotensive. Cocaine-induced catecholamine effects can clinically mimic pheochromocytoma, thereby complicating the diagnosis. In addition, the biologic behavior of CP with neuroblastoma is uncertain and warrants oncologic assessment and surveillance.
Keywords: Composite pheochromocytoma; Neuroblastoma; Adrenal
| Introduction | ▴Top |
Composite pheochromocytoma (CP) is a very rare tumor of the adrenal medulla, consisting of both neuroendocrine and neural components. Histologically, the endocrine portion is that of a pheochromocytoma, an uncommon tumor arising from the catecholamine-producing chromaffin cells of the adrenal medulla, while the neural elements have been reported as neuroblastoma, ganglioneuroma, ganglioneuroblastoma, neuroendocrine carcinoma, and, rarely, Schwannoma [1]. Although both pheochromocytomas and neurogenic tumors are derived from the neural crest, they are distinct entities. Symptoms can result from hypersecretion of hormones by either component [2]. However, not all patients present with classic symptoms and medical conditions; medications and illicit drugs can mimic these classic symptoms and/or confound commonly used screening tests.
We report a case of a patient with classic symptoms of pheochromocytoma who concomitantly used cocaine, and we discuss challenges encountered during the diagnostic and treatment courses.
| Case Report | ▴Top |
A 46-year-old Caucasian man was seen in the endocrinology clinic because of a 2-year history of intermittent headaches, palpitations and diaphoresis. These episodes would occur every 2 - 3 days and last up to 20 min, and were associated with pallor, nausea and vomiting, and blurry vision. He denied any chest pain, loss of consciousness or diarrhea.
He had a history of a remote tonsillectomy and hypothyroidism, for which he had been taking levothyroxine, 0.050 mg daily. There were no known drug allergies. The patient was single and unemployed, had a 15 pack year history of smoking cigarettes, smoked marijuana every 2 - 3 days and snorted cocaine intermittently. He consumed three alcoholic beverages per week. There was no family history of pheochromocytoma, hyperparathyroidism or thyroid malignancies.
On examination, the patient was a thin man with initial supine and standing blood pressures of 140/90 and 140/80 mmHg, respectively. Supine and standing pulses were 92 and 96 beats/min, respectively. While in the clinic, the patient had a hypertensive episode with tachycardia (blood pressure 170/90 mmHg and heart rate 100 beats/min). He was not diaphoretic or pale during that episode. The rest of the physical examination was unremarkable.
The 24-h urine levels of catecholamines, metanephrines and vanillylmandelic acid (VMA) are summarized in Table 1. Though all analytes were above the reference range, the urinary epinephrine and metanephrine were the most markedly elevated, at approximately seven and ten times the upper limit of normal, respectively. A magnetic resonance imaging (MRI) of the brain, performed a year earlier for complaints of headaches, demonstrated nonspecific white matter changes.
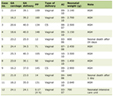 Click to view | Table 1. Initial 24-H Urine for Fractionated Catecholamines and Metanephrines |
Although this patient’s presentation was suspicious for pheochromocytoma, it was difficult to differentiate this from cocaine-induced catecholamine effects. Another consideration was that this patient had superimposed anxiety and panic attacks. In order to ensure that the elevated urinary catecholamines and metanephrines were not the result of cocaine use (which is known to increase urinary catecholamine levels), the plan was to repeat the urine collection along with a urinary toxicology screen. Unfortunately, the patient did not repeat the collection and was lost to follow-up.
Two years later, the patient was seen by a neurologist for “full body trembling”, which had been occurring for the past few years, and was not accompanied by any impairment of consciousness. An electroencephalogram (EEG) was unremarkable. The neurologist felt that these episodes were related to cocaine-induced catecholamine surges rather than seizure activity. Abdominal CT revealed a 5.8 × 5 cm mass in the left adrenal gland, with an attenuation of approximately 40 HU (Hounsfield units), highly suspicious for pheochromocytoma (Fig. 1). No calcifications or adenopathy were seen, and the contralateral adrenal gland was unremarkable. There was a 1.8 cm hypodensity in segment 8 of the liver, which was found to be consistent with a hemangioma on subsequent ultrasound correlation.
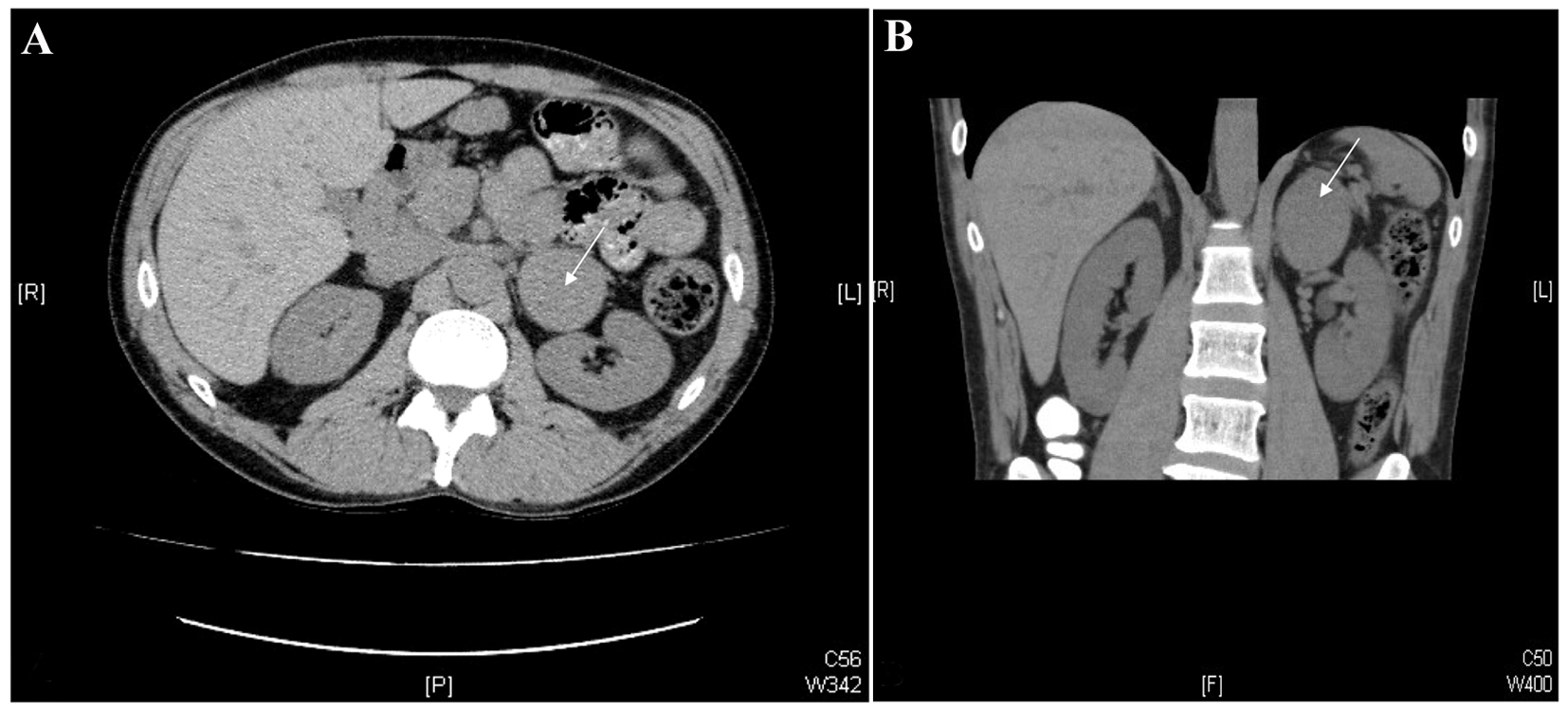 Click for large image | Figure 1. CT images of the abdomen. Axial (A) and coronal (B) images from a non-contrast-enhanced CT scan of the abdomen. Note the 5.8× 5.0 cm mass in the left adrenal gland (white arrows). |
Several weeks after the CT scan, the patient presented to the emergency room with another paroxysm. His initial blood pressure was as high as 187/122 mmHg with a heart rate of 85 beats/min. His blood pressure later spontaneously dropped to approximately 100 mmHg systolic. At that time, he was started on prazosin 1 mg po tid and told to liberalize his sodium intake. He was also referred back to his original endocrinologist for further management of his pheochromocytoma.
When reassessed in the endocrinology clinic, he endorsed a 25-pound weight loss over the previous 4 years despite a voracious appetite. The remainder of his history was unchanged from the initial assessment. On examination, his weight was 68 kg. Blood pressure in the supine position was 144/76 mmHg with a heart rate of 100 beats/min. In the standing position, his blood pressure was 124/90 mmHg with a heart rate of 108 beats/min. Dermatologic examination revealed two cafe-au-lait spots: one in his abdomen, and another one on his back. He had one axillary freckle bilaterally, as well as an intradermal cyst in his upper back. No neurofibromas were seen, nor were there any mucosal neuromas in his oral cavity or conjunctivae.
The patient was started on bisoprolol 2.5 mg daily while arrangements were being made for resection of the pheochromocytoma. He was asked to repeat his 24-h urine collection for catecholamines and metanephrines while abstaining from cocaine; however, the patient was unable to comply with the urine collection. Other than occasional headaches, he had no further spells of tremors, palpitations or diaphoresis. Two weeks prior to surgery, the patient was started on phenoxybenzamine 10 mg bid, at which time the prazosin was discontinued, resulting in good control of his hypertension. Plasma free metanephrines and chromogranin A were markedly elevated (Table 2).
Click to view | Table 2. Preoperative Plasma Free Metanephrines and Chromogranin A |
He underwent an uncomplicated laparoscopic left adrenalectomy. There were no findings to suggest a metastatic neoplasm. He did have some runs of hypertension intraoperatively that were effectively controlled with medical therapy. His post-operative course was uneventful, antihypertensive medications were discontinued and the patient remained normotensive.
The enlarged adrenal weighed 80 g and measured 7 × 5.5 × 4.5 cm. The organ was extensively replaced by a solid, homogenous brown tumor that was confined to the adrenal. There was no cystic change or calcification evident grossly or microscopically. Histological examination showed a tumor replacing the medulla with a thin rim of residual cortex, the microscopic features of which were a typical pheochromocytoma (Fig. 2A). In some areas, there were infiltrates of small blue cells, confirmed as neuroblastoma with NB84a (anti-neuroblastoma antibody) immunoperoxidase stain (Fig. 2B). The mitosis karyorrhexis index (MKI) was low and there was no ganglioneuromatous infiltrate evident (Fig. 2C). The neuroblastoma component comprised about 5% of the overall tumor volume. In light of these findings, additional sections were submitted and the gross specimen was re-examined; there were no areas that could be identified as different from the typical pheochromocytoma.
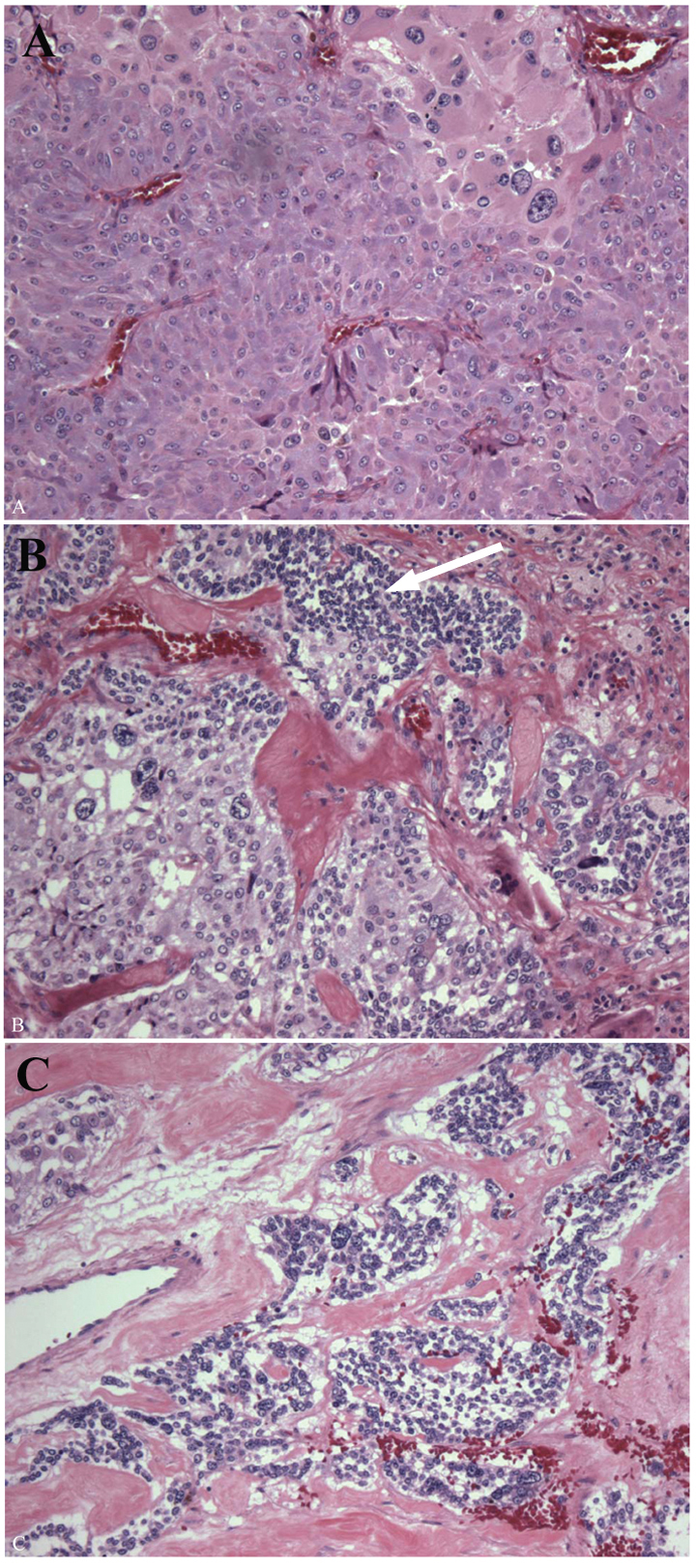 Click for large image | Figure 2. Pathology of the left adrenal tumor. The enlarged adrenal gland (80 g) was replaced by a homogenous red brown tumor, the majority of which was a typical pheochromocytoma composed of polygonal cells with abundant eosinophilic cytoplasm and scattered enlarged bizarre cells (A). Pheochromocytoma merged with microscopic foci of small round blue cells (white arrow), these comprising < 5% of the tumor and not evident grossly (B). The nests of immature neuroblast cells showed a low MKI (C). |
A CT chest was performed within the ensuing 6 weeks, which revealed no evidence of recurrent or metastatic disease, and a subsequent bone scan was negative. Regrettably, he did not attend his scheduled CT abdomen or I-123 metaiodobenzylguanidine (MIBG), nor did he comply with biochemical evaluation.
He was assessed by medical oncology in view of the diagnosis of composite pheochomocytoma with neuroblastomatous elements, and given the fact that his disease was completely resected, and the volume of neuroblastoma with a low MKI, no adjuvant systemic therapy was recommended.
The patient was seen in the endocrinology clinic 6 months postoperatively. He was feeling quite well, and denied any further paroxysms of palpitations, diaphoresis or headaches. His blood pressure was 112/77 mmHg with a heart rate of 56 beats/min. The remainder of his examination was unchanged. He remains asymptomatic one year postoperatively.
| Discussion | ▴Top |
CP is a very rare tumor that accounts for only 3% of both adrenal and extra-adrenal pheochromocytomas [1]. Less than 50 cases of CP have been reported in the English literature [2]. Most cases have been associated with ganglioneuromas - the occurrence with neuroblastoma is infrequent and the clinical demographics have not been well elucidated because of its rarity.
Both pheochromocytoma and neuroblastoma arise from the neural crest, but differ in their features. Pheochromocytomas are rare benign tumors that occur mainly in adults between the ages of 30 and 50 years [3-6]. They arise from the chromaffin tissue and produce catecholamines [7], typically causing sustained hypertension or paroxysmal hypertensive episodes, along with the signs and symptoms of excessive adrenergic effects [8, 9]. On the other hand, neuroblastoma, a malignancy of sympathetic ganglia, tends to occur in childhood: approximately 90% of patients present before 5 years of age [10]. Adult neuroblastomas are extremely rare with an annual incidence of 0.12 cases per million (US data 1998-2002) [11] and hence most adult patients are treated according to pediatric guidelines [12].
The nonspecific clinical manifestations of CP [2] and rarity of the combination with neuroblastoma [13] may delay diagnosis in the absence of clinical suspicion. CP can cause symptoms related to hormonal secretion by one or both components of the tumor [14]. Elevated plasma or urinary dopamine or homovanillic acid (HVA) in a patient with suspected pheochromocytoma may suggest the presence of an additional neural component [15]. However, HVA levels are influenced by the size of the neuroblastoma and there are similarities between markers. Therefore, clinical features - ranging from a silent course to a vasoactive intestinal peptide (VIP)-mediated watery diarrhea syndrome in ganglioneuromatous CP [16, 17] - and serological tests are unlikely to diagnose the neuroblastoma component.
Additionally, functional pheochromocytoma may mimic panic disorders, hyperthyroidism, and illicit drug use, among other conditions, thereby hindering the diagnosis [18, 19]. Although the pheochromocytoma component of our patient’s CP was clearly functional, the history of cocaine use, which can increase urinary catecholamines and masquerade as pheochromocytoma [18], was a complicating factor, as was the absenteeism at follow-up. Nevertheless, on the subsequent presentation, the history alerted the team to the possibility of there being an underlying pheochromocytoma, prompting further radiological investigation.
The high attenuation (40 HU) and well-circumscribed homogenous appearance seen on our patient’s CT are similar to what would be expected for a classic pheochromocytoma, and therefore the level of suspicion for a CP or any malignant component was low [20-22]. It should be noted, though, that radiologic features are neither specific nor diagnostic for CP [23].
Generally, the diagnosis of CP is made on microscopy, where various admixtures of pheochromocytoma and neural components are seen [2, 24]. In approximately 70% of CPs, the neural component is found to be ganglioneuroma [24]. The NB84a monoclonal antibody is a useful marker confirming the diagnosis of a neuroblastoma [25].
Complete surgical resection with careful preoperative alpha-adrenergic blockade and subsequent beta-adrenergic blockade for any consequent tachyarrhythmia [26], in the presence of a functional pheochromocytoma, is the most appropriate therapy for CP with neuroblastoma. Prazosin is a commonly used selective alpha-1 adrenergic agonist, but requires frequent dosing due to a short half-life and its serum concentration may decrease to ineffective levels at the time of surgery if used preoperatively [27]. In contrast, phenoxybenzamine, a non-competitive, non-selective alpha-blocker, has a longer half-life of over 24 h [28].
A laparoscopic approach to adrenalectomy has been associated with reduced postoperative morbidity, shorter hospital stays [28] and stable intraoperative blood pressures, presumably due to reduced tumor handling [29]. To ensure complete and successful resection of the pheochromocytoma component, testing for free plasma or fractionated urinary metanephrines is indicated 2 - 6 weeks after surgery [30, 31]. Normal values are considered to be evidence of tumor eradication, but high postoperative metanephrine levels suggest persistent disease and warrant further imaging [31].
Although there is no clear consensus for treating adult neuroblastoma, complete surgical resection is sufficient for low risk and non-metastatic tumors, but combination therapy with chemotherapy and radiotherapy is needed for metastatic and recurrent disease [32]. The treatment paradigm for these tumors is poorly understood because of their rarity and variations in biologic behavior [33]. Adjuvant therapy was not advised for our patient because he was deemed to be at lower risk based on the low volume neuroblastomatous component and complete resection of the tumor that was confined to the adrenal.
There has been little literature on long-term follow-up of patients with CP and the clinical course of these rare tumors has not been well established. In their review article, Amar et al proposed long-term follow-up recommendations for classic pheochromocytoma and paraganglioma [31]. In cases with large tumors and normal or unmeasured preoperative metanephrines, imaging within 3 months is recommended [31]. For pheochromocytomas > 5 cm in diameter, as well as familial pheochromocytomas and paragangliomas, clinical and biochemical assessment 6 months postoperatively and annually thereafter is recommended. For smaller pheochromocytomas (≤ 5 cm), an initial follow-up at 12 months and then every alternate year thereafter has been proposed [31]. Assessment for adrenergic symptoms and blood pressure monitoring are essential [14, 31].Schalk et al also recommended monitoring urine for VMA and HVA in CPs given the risk of recurrence and metastasis, posed by the presence of the neuroblastoma component [32].
Any adrenergic signs or symptoms warrant earlier biochemical testing, and if positive, imaging with CT or MRI of the thorax, abdomen and pelvis to rule out any recurrence [31]. Imaging has an important role for the detection of nonfunctional tumors, and positron emission tomography with [18F] fluoro-2-deoxy-D-glucose ([18F]FDG-PET) can be helpful to distinguish new tumors from metastases and for tumor staging [31].
Recurrence rates for pheochromocytomas are reported to be 14% and can occur up to 16 years after surgery [30]. For CPs, prognosis is variable. Metastases from both components are reported, but mainly from the neural component [14]. Overall, the malignancy rate for CP is approximately 25% [2] and due to the risk of recurrence and metastasis, the presence of neuroblastoma is an indicator of poor prognosis; however, the presence of neural components does not always imply a poor prognosis [2, 15].
Neuroblastomas in adults tend to present at a more advanced stage than in children and may have a more aggressive phenotype, with ultimate survival being less than 50% [11, 12]. The prognosis depends on numerous variables, the most important being age at diagnosis, grade and stage of the tumor [34]. In children with neuroblastoma, an increased amplification of the oncogene N-myc is a marker for poor outcome [11, 12, 35]; however, it tends to be unamplified in adults with neuroblastoma. Also, neither CP nor classic pheochromocytomas display N-myc amplification [35].
The clinical significance of microscopic detection of the nonpheochromocytoma component of CPs has yet to be clarified, due to the paucity of reports. However, the identification of composite tumors is important because it adds to our accumulation of cases and can be used to help predict the biologic behavior of CP [24]. A case series by Comstock et al compared four cases of CP with selected cases of ordinary pheochromocytoma and neuroblastoma [35]. Following adrenalectomy, none of the patients developed recurrences or metastases at a mean follow-up of 4.9 years, suggesting that CP may be regarded as a histologic variant of classic pheochromocytoma [14]. It should be noted, however, that this was a very small sample size, and therefore these cases may not necessarily be generalizable to all CPs.
The underlying cause of CP in our patient has yet to be determined. Twenty-three percent of CPs are reportedly associated with NF1, an autosomal dominant disorder [36]. The features of NF1 include benign tumors of the peripheral nervous system and an increased risk of developing malignancies. Pheochromocytomas occur in 1% of patients with NF1 [37], and 23% are CPs associated with NF1 [38]. The latter has a variable presentation. Our patient had cafe-au-lait spots and axillary freckling, both of which are hallmarks of NF1. However, the diagnostic criteria for NF1 require the presence of neurofibromas [38], which our patient lacked. Although these findings in the absence of a positive family history and late presentation would make a diagnosis of NF1 unlikely, de novomutations in NF1 are reported without neurofibromas [39]. Furthermore, new research suggests that 15-25% of apparently sporadic pheochromocytomas have mutations in MEN (RET), NF1, or VHL genes [7, 40]. Our patient has been referred to the Clinical Genetics Program for further assessment.
Conclusions
This is a very rare case of a CP involving a pheochromocytoma combined with a neuroblastoma. Since its signs and symptoms are common to many other pathological conditions, the diagnosis of pheochromocytoma can be quite challenging. However, due to the potentially devastating consequences of an undiagnosed pheochromocytoma, the clinician must maintain a high index of suspicion when patients present with compatible signs and/or symptoms. Our patient’s concomitant cocaine use added to the diagnostic complexity, as cocaine is known to increase urinary catecholamines and clinically can mimic a pheochromocytoma.
Our patient is the sixth reported case of CP with neuroblastoma [13, 35, 41, 42]. The finding of a neuroblastomatous infiltrate requires further pathologic assessment to determine the volume of the immature component and risk factors such as MKI. The biologic behavior of these lesions is uncertain and warrants oncologic assessment and follow-up.
Disclosure
The authors have nothing to declare.
Author Contributions
OS carried out the clinical management of the patient and participated in the drafting of the manuscript. JF participated in the drafting of the manuscript. JR examined and photographed the pathology specimen, and participated in the critical revision of the manuscript. AP carried out the clinical management of the patient and participated in the critical revision of the manuscript. All authors read and approved the final manuscript.
Abbreviations
CP: composite pheochromocytoma; VMA: vanillylmandelic acid; MRI: magnetic resonance imaging; EEG: electroencephalogram; CT: computed tomography; HU: Hounsfield units; NB84a: anti-neuroblastoma antibody; MKI: mitosis karyorrhexis index; MIBG: metaiodobenzylguanidine; MEN: multiple endocrine neoplasia; NF1: neurofibromatosis type 1; VHL: von Hippel-Lindau; HVA: homovanilic acid; VIP: vasoactive intestinal peptide; FDG-PET: fluorodeoxyglucose positron emission tomography
| References | ▴Top |
- Menon S, Mahajan P, Desai SB. Composite adrenal medullary tumor: A rare cause of hypertension in a young male. Urol Ann. 2011;3(1):36-38.
doi pubmed - Khan AN, Solomon SS, Childress RD. Composite pheochromocytoma-ganglioneuroma: a rare experiment of nature. Endocr Pract. 2010;16(2):291-299.
doi pubmed - Beard CM, Sheps SG, Kurland LT, Carney JA, Lie JT. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin Proc. 1983;58(12):802-804.
pubmed - Young WF, Jr. Management approaches to adrenal incidentalomas. A view from Rochester, Minnesota. Endocrinol Metab Clin North Am. 2000;29(1):159-185, x.
doi - Rha SE, Byun JY, Jung SE, Chun HJ, Lee HG, Lee JM. Neurogenic tumors in the abdomen: tumor types and imaging characteristics. Radiographics. 2003;23(1):29-43.
doi pubmed - Amar L, Bertherat J, Baudin E, Ajzenberg C, Bressac-de Paillerets B, Chabre O, Chamontin B, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. 2005;23(34):8812-8818.
doi pubmed - Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366(9486):665-675.
doi - Calhoun DA, Jones D, Textor S, Goff DC, Murphy TP, Toto RD, White A, et al. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension. 2008;51(6):1403-1419.
doi pubmed - Strosberg JR. Update on the management of unusual neuroendocrine tumors: pheochromocytoma and paraganglioma, medullary thyroid cancer and adrenocortical carcinoma. Semin Oncol. 2013;40(1):120-133.
doi pubmed - Park JR, Eggert A, Caron H. Neuroblastoma: biology, prognosis, and treatment. Hematol Oncol Clin North Am. 2010;24(1):65-86.
doi pubmed - Esiashvili N, Goodman M, Ward K, Marcus RB, Jr., Johnstone PA. Neuroblastoma in adults: Incidence and survival analysis based on SEER data. Pediatr Blood Cancer. 2007;49(1):41-46.
doi pubmed - Franks LM, Bollen A, Seeger RC, Stram DO, Matthay KK. Neuroblastoma in adults and adolescents: an indolent course with poor survival. Cancer. 1997;79(10):2028-2035.
doi - Candanedo-Gonzalez FA, Alvarado-Cabrero I, Gamboa-Dominguez A, Cerbulo-Vazquez A, Lopez-Romero R, Bornstein-Quevedo L, Salcedo-Vargas M. Sporadic type composite pheochromocytoma with neuroblastoma: clinicomorphologic, DNA content and ret gene analysis. Endocr Pathol. 2001;12(3):343-350.
doi - Lau SK, Chu PG, Weiss LM. Mixed cortical adenoma and composite pheochromocytoma-ganglioneuroma: an unusual corticomedullary tumor of the adrenal gland. Ann Diagn Pathol. 2011;15(3):185-189.
doi pubmed - Fujiwara T, Kawamura M, Sasou S, Hiramori K. Results of surgery for a compound adrenal tumor consisting of pheochromocytoma and ganglioneuroblastoma in an adult: 5-year follow-up. Intern Med. 2000;39(1):58-62.
doi pubmed - George DJ, Watermeyer GA, Levin D, Epstein D, Ross IL, Scholz BU, Setshedi M, et al. Composite adrenal phaeochromocytoma-ganglioneuroma causing watery diarrhoea, hypokalaemia and achlorhydria syndrome. Eur J Gastroenterol Hepatol. 2010;22(5):632-634.
doi pubmed - Mahajan H, Lee D, Sharma R, Chin P, Watt WH, McBride G, Bilous M. Composite phaeochromocytoma-ganglioneuroma, an uncommon entity: report of two cases. Pathology. 2010;42(3):295-298.
doi pubmed - Koneth I, Suter PM, Vetter W. [Arterial hypertension and increased urinary catecholamines]. Praxis (Bern 1994). 2000;89(31-32):1252-1256.
- Mena A, Lawson M, Kabadi UM. Pheochromocytoma. Endocr Pract. 1997;3(2):98-105.
doi pubmed - Jacques AE, Sahdev A, Sandrasagara M, Goldstein R, Berney D, Rockall AG, Chew S, et al. Adrenal phaeochromocytoma: correlation of MRI appearances with histology and function. Eur Radiol. 2008;18(12):2885-2892.
doi pubmed - Blake MA, Krishnamoorthy SK, Boland GW, Sweeney AT, Pitman MB, Harisinghani M, Mueller PR, et al. Low-density pheochromocytoma on CT: a mimicker of adrenal adenoma. AJR Am J Roentgenol. 2003;181(6):1663-1668.
doi pubmed - Kalra MK, Blake MA, Boland GW, Hahn PF. CT features of adrenal pheochromocytomas: attenuation value and loss of contrast enhancement. Radiology. 2005;236(3):1112-1113.
doi pubmed - Lam KY, Lo CY. Composite Pheochromocytoma-Ganglioneuroma of the Adrenal Gland: An Uncommon Entity with Distinctive Clinicopathologic Features. Endocr Pathol. 1999;10(4):343-352.
doi - Kino NR, Kim T, Lee JN et al. Laparoscopically resected composite pheochromocytoma ganglioneuroma. Endocrinol Metab (Seoul). 2011;26(4):340-344.
doi - Miettinen M, Chatten J, Paetau A, Stevenson A. Monoclonal antibody NB84 in the differential diagnosis of neuroblastoma and other small round cell tumors. Am J Surg Pathol. 1998;22(3):327-332.
doi pubmed - Mannelli M, Dralle H, Lenders JW. Perioperative management of pheochromocytoma/paraganglioma: is there a state of the art? Horm Metab Res. 2012;44(5):373-378.
doi pubmed - Kinney MA, Warner ME, vanHeerden JA, Horlocker TT, Young WF, Jr., Schroeder DR, Maxson PM, et al. Perianesthetic risks and outcomes of pheochromocytoma and paraganglioma resection. Anesth Analg. 2000;91(5):1118-1123.
pubmed - Ramachandran MS, Reid JA, Dolan SJ, Farling PA, Russell CF. Laparoscopic adrenalectomy versus open adrenalectomy: results from a retrospective comparative study. Ulster Med J. 2006;75(2):126-128.
pubmed - Kercher KW, Novitsky YW, Park A, Matthews BD, Litwin DE, Heniford BT. Laparoscopic curative resection of pheochromocytomas. Ann Surg. 2005;241(6):919-926; discussion 926-918.
- Darr R, Lenders JW, Hofbauer LC, Naumann B, Bornstein SR, Eisenhofer G. Pheochromocytoma - update on disease management. Ther Adv Endocrinol Metab. 2012;3(1):11-26.
doi pubmed - Amar L, Fassnacht M, Gimenez-Roqueplo AP, Januszewicz A, Prejbisz A, Timmers H, Plouin PF. Long-term postoperative follow-up in patients with apparently benign pheochromocytoma and paraganglioma. Horm Metab Res. 2012;44(5):385-389.
doi pubmed - Schalk E, Mohren M, Koenigsmann M, Buhtz P, Franke A, Jentsch-Ullrich K. Metastatic adrenal neuroblastoma in an adult. Onkologie. 2005;28(6-7):353-355.
doi pubmed - Rougier P, Mitry E. Chemotherapy in the treatment of neuroendocrine malignant tumors. Digestion. 2000;62(Suppl 1):73-78.
doi pubmed - Cotterill SJ, Pearson AD, Pritchard J, Foot AB, Roald B, Kohler JA, Imeson J. Clinical prognostic factors in 1277 patients with neuroblastoma: results of The European Neuroblastoma Study Group 'Survey' 1982-1992. Eur J Cancer. 2000;36(7):901-908.
doi - Comstock JM, Willmore-Payne C, Holden JA, Coffin CM. Composite pheochromocytoma: a clinicopathologic and molecular comparison with ordinary pheochromocytoma and neuroblastoma. Am J Clin Pathol. 2009;132(1):69-73.
doi pubmed - Dluhy RG. Pheochromocytoma—death of an axiom. N Engl J Med. 2002;346(19):1486-1488.
doi pubmed - Kimura N, Watanabe T, Fukase M, Wakita A, Noshiro T, Kimura I. Neurofibromin and NF1 gene analysis in composite pheochromocytoma and tumors associated with von Recklinghausen's disease. Mod Pathol. 2002;15(3):183-188.
doi pubmed - Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 2009;61(1):1-14; quiz 15-16.
doi pubmed - Quintans B, Beleza S, Brion M, Sanchez-Diz P, Lareu M, Carracedo A. Population data of Galicia (NW Spain) on the new Y-STRs DYS437, DYS438, DYS439, GATA A10, GATA A7.1, GATA A7.2, GATA C4 and GATA H4. Forensic Sci Int. 2003;131(2-3):220-224.
doi - Benn DE, Robinson BG. Genetic basis of phaeochromocytoma and paraganglioma. Best Pract Res Clin Endocrinol Metab. 2006;20(3):435-450.
doi pubmed - Tatekawa Y, Muraji T, Nishijima E, Yoshida M, Tsugawa C. Composite pheochromocytoma associated with adrenal neuroblastoma in an infant: a case report. J Pediatr Surg. 2006;41(2):443-445.
doi pubmed - Franquemont DW, Mills SE, Lack EE. Immunohistochemical detection of neuroblastomatous foci in composite adrenal pheochromocytoma-neuroblastoma. Am J Clin Pathol. 1994;102(2):163-170.
pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.