

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website http://www.jofem.org |
Case Report
Volume 5, Number 5, October 2015, pages 309-311
Neurofibromatosis Type 1 With a Pheochromocytoma: A Rare Presentation of Von Recklinghausen Disease
Wahib Zafara, Benjamin Chaucera, b, Fidencio Davalosa, Siddiqui Beenisha, Marie Chevenona, Jay Nfonoyima
aRichmond University Medical Center, 355 Bard Avenue, Staten Island, NY 10310, USA
bCorresponding Author: Benjamin T. Chaucer, Richmond University Medical Center, 400 Forest Avenue Apt 2R, Staten Island, NY 10301, USA
Manuscript accepted for publication September 23, 2015
Short title: Neurofibromatosis Type 1
doi: http://dx.doi.org/10.14740/jem308w
| Abstract | ▴Top |
The combination of Von Recklinghausen disease with a pheochromocytoma has a genetic linkage but is an exceedingly rare occurrence. Von Recklinghausen disease or neurofibromatosis type 1 (NF1) is a neurocutaneous disorder characterized by cafe-au-lait macules and multiple, soft tissue neurofibromas. While a pheochromocytoma is a neuroendocrine tumor characterized by intermittent hypertension. NF1 predisposes patients to pheochromocytomas; however, this occurrence is extremely rare. We describe the case of a young male with neurofibromatosis and a pheochromocytoma.
Keywords: Pheochromocytoma; Neurofibromatosis; Von Recklinghausen disease
| Introduction | ▴Top |
We present the case of neurofibromatosis with a pheochromocytoma. This is a rare presentation of Von Recklinghausen disease. While it is known that neurofibromatosis type 1 (NF1) predisposes patients to pheochromocytomas, this represents a unique presentation [1]. This is rarely seen in clinical practice.
| Case Report | ▴Top |
A 32-year-old male of Azakaneshi Jewish descent, previously diagnosed with NF1, presented with nausea, vomiting and weight loss. His past medical history included hypercholesterolemia and DM. His family history was positive for his mother having NF1. The social history was negative for smoking, alcohol and recreational drug use. On review of systems, patient complained of nausea and vomiting. He denied any phobias, anxiety, panic attacks, palpitations, urine urgency or frequency, headaches, abdominal pain, cramping or bloating. Examination of the skin revealed cafe-au-lait spots and cutaneous neurofibromas. No flushing of the skin was seen, and mild pallor was noted. The heart sounds were normal, without murmurs, or elevation of venous pressure. The blood pressure ranged from 140/75 to 225/110 mm Hg with a persistent tachycardia reaching a maximum of 125 beats per minute. There was no wheezing and breath sounds were equal bilaterally. There was no abdominal tenderness, guarding or pain upon palpation. Given the patient’s history of NF1, a pheochromocytoma needed to be excluded. The patient’s thyroid function tests were within normal limits. The patients initial symptoms of nausea and vomiting resolved with blood pressure control.
The electrocardiogram showed only sinus tachycardia. Abdominal MRI demonstrated a left adrenal gland within normal limits. A mass was found on the right adrenal gland measuring 7.2 × 5.6 cm. It was hypervascular in nature, and exerted a mass effect on upper pole of the right kidney. A PET scan using fluorodeoxyglucose showed the adrenal mass to be not significantly active with no evidence of metastasis. Biochemical testing showed elevated levels of serum and urine catecholamines, metanephrines and vanillylmandelic acid (Table 1 and 2). The clinical diagnosis of pheochromocytoma was confirmed pathologically after the mass was surgically removed.
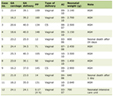 Click to view | Table 1. Plasma Concentration of Metabolites |
Click to view | Table 2. 24-Hour Urinary Metabolites Level |
| Discussion | ▴Top |
Neurofibromatosis is an autosomal dominant disease that was first described by Von Recklinghausen in 1849. It is characterized by multiple soft tissue neurofibromas, cafe-au-lait macules, axillary and inguinal freckling, iris hamartomas (Lisch nodules), bony abnormalities, CNS gliomas, macrocephaly, and cognitive deficits [2]. NF1 results from mutation of the NF1 gene, a tumor suppressor found on chromosome 17q11.2 that leads to its inactivation [3]. This predisposes affected individuals to develop a number of neoplasms that most commonly affect the tissues of the eye, skin and nervous system. In a rare subset of patients, it also predisposes to a number of less common tumors of neuroectodermal or mesenchymal origin, including pheochromocytoma [4].
Pheochromocytoma is a neuroendocrine tumor of the adrenal medulla or the extra-adrenal chromaffin tissue that failed to involute after birth. The tumor produces excessive amounts of catecholamine resulting in tachycardia and hypertension [5]. This clinical picture was first recognized by Feil Frankel in 1886. The condition exists either as a sporadic or familial type, the latter of which is seen in NF1. The NF1 gene is a tumor suppressor gene mapping to chromosome 17q11.2 [6], but the gene responsible for the specific association between NF1 and pheochromocytoma is still unknown [7].
Pheochromocytoma is found in 0.1% among those tested because of hypertension and in 4% of patients with adrenal incidentaloma. The incidence in the general population is estimated to be 1 per 100,000 persons per year or less. They may be classified as sporadic or familial. Most of the pheochromocytomas are sporadic. Familial pheochromocytomas are seen mainly in patients with multiple endocrine neoplasia type 2, NF1, von Hippel-Lindau disease and familial carotid body tumors [6]. NF1 affects approximately 1 in 3,500 patients worldwide. Pheochromocytoma occurs in 0.1-5.7% of patients with NF1, but the incidence increases to 20-50% in cases of NF1 with hypertension compared to 0.1% of all hypertensive individuals [6]. Pheochromocytoma and ganglia neuroblasts arise from a common precursor deriving from the primitive neuroectoderm or the neural crest. NF1 is also a dysplastic disease of the neuroectoderm. Therefore, an association between NF1 and pheochromocytoma has been described, although its incidence is very low.
NF1-associated pheochromocytoma has germ line NF1 mutations that favor the cysteine serine-rich domain [8]. This behaves like a sporadic pheochromocytoma. They predominantly secrete epinephrine and present with classical episodic symptoms like palpitations, diaphoresis, headaches, panic attacks and paroxysmal hypertension. Persistent hypertension is seen in about 60% of cases of NF1 [9] with pheochromocytoma as seen in our patient. The tumors are generally localized to adrenals and are rarely malignant. Approximately 10-15% of catecholamine-secreting tumors are extra-adrenal and are referred to as catecholamine-secreting paragangliomas. About 10% of pheochromocytomas are bilateral [10]. About 10% of all catecholamine-secreting tumors are malignant; however, this cannot be determined at the time of diagnosis. Malignant pheochromocytomas are histologically and biochemically the same as benign ones. The only reliable clue to the presence of a malignant pheochromocytoma is local invasion into surrounding tissues and organs or distant metastases. This may occur as long as 20 years after resection. Many malignant pheochromocytomas are found discovered to be so due to recurrence after a complete gross resection [11]. According to the WHO, malignancy is defined in the presence of metastasis. Thus, even when pheochromocytomas or paragangliomas are considered “benign” on pathologic examination, long term follow-up is indicated in all patients. The presence of tumor necrosis, Ki-67 index > 4% and pS100 absence indicate a high risk of malignancy/recurrence. Our patient’s pathology report had absence of tumor necrosis, pS100 and a Ki-67 index of 2%. The mean age at diagnosis of pheochromocytoma in patients with NF1 is 42 years, with most occurring between the third to fourth decade of life. In these patients, the catecholamine-secreting tumor is usually a solitary benign adrenal pheochromocytoma. Persons with NF1 are at increased risk for malignant conditions, especially malignant peripheral nerve sheath tumor (MPNST), leukemia and rhabdomyosarcoma.
Adequate preoperative preparation is required with adrenergic blockers and surgery offers best cure rate [12]. Postoperative complications include hypotension and hypoglycemia. A long-term follow-up is required to monitor for recurrence in patients with genetic predisposition.
Conclusion
Our patient had a rare combination of NF1 with a pheochromocytoma. It was further unique because our patient did not present with the classical features of pheochromocytoma. Data from observational studies suggest that all patients with NF1 and hypertension should undergo a biochemical testing for pheochromocytoma [11]. A high index of suspicion is required for identification of such a rare association.
| References | ▴Top |
- Walther MM, Herring J, Enquist E, Keiser HR, Linehan WM. von Recklinghausen's disease and pheochromocytomas. J Urol. 1999;162(5):1582-1586.
doi - Dannenberg H, van Nederveen FH, Abbou M, Verhofstad AA, Komminoth P, de Krijger RR, Dinjens WN. Clinical characteristics of pheochromocytoma patients with germline mutations in SDHD. J Clin Oncol. 2005;23(9):1894-1901.
doi pubmed - Pacak K, Linehan WM, Eisenhofer G, Walther MM, Goldstein DS. Recent advances in genetics, diagnosis, localization, and treatment of pheochromocytoma. Ann Intern Med. 2001;134(4):315-329.
doi pubmed - Zografos GN, Vasiliadis GK, Zagouri F, Aggeli C, Korkolis D, Vogiaki S, Pagoni MK, et al. Pheochromocytoma associated with neurofibromatosis type 1: concepts and current trends. World J Surg Oncol. 2010;8:14.
doi pubmed - Erlic Z, Rybicki L, Peczkowska M, Golcher H, Kann PH, Brauckhoff M, Mussig K, et al. Clinical predictors and algorithm for the genetic diagnosis of pheochromocytoma patients. Clin Cancer Res. 2009;15(20):6378-6385.
doi pubmed - Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346(19):1459-1466.
doi pubmed - Pawlu C, Bausch B, Reisch N, Neumann HP. Genetic testing for pheochromocytoma-associated syndromes. Ann Endocrinol (Paris). 2005;66(3):178-185.
doi - Shen MH, Harper PS, Upadhyaya M. Molecular genetics of neurofibromatosis type 1 (NF1). J Med Genet. 1996;33(1):2-17.
doi pubmed - Welander J, Soderkvist P, Gimm O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer. 2011;18(6):R253-276.
doi pubmed - Datta Saikat, Sharmistha Bhattacherjee. Type I Neurofibromatosis With Pheochromocytoma. Int J Med Sci Public Health. 2013;2(3):630.
doi - Chen H, Sippel RS, O'Dorisio MS, Vinik AI, Lloyd RV, Pacak K. The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010;39(6):775-783.
doi pubmed - IYODA, Akira et al. A Case Of Pheochromocytoma And Paraganglioma With Family History. The Journal of the Japanese Practical Surgeon Society. 1994;55(12):3195-3200.
doi
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.