

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website https://www.jofem.org |
Original Article
Volume 12, Number 3, June 2022, pages 79-88

Growth Hormone Deficiency in Childhood Intracranial Germ Cell Tumor Survivors
Diana W. Lonea, b, c, g ![]() , Karim T. Sadaka, d, Bradley S. Millera, d
, Karim T. Sadaka, d, Bradley S. Millera, d ![]() , Jeannette M. Sampleb, Aubrey K. Hubbardb, Caryn Wolterb, Michelle Roeslerd, Michelle Nunoe, f
, Jeannette M. Sampleb, Aubrey K. Hubbardb, Caryn Wolterb, Michelle Roeslerd, Michelle Nunoe, f ![]() , Jenny N. Poynterb, d
, Jenny N. Poynterb, d ![]()
aDepartment of Pediatrics, University of Minnesota, MN 55455, USA
bDivision of Epidemiology and Clinical Research, Department of Pediatrics, University of Minnesota, Minneapolis, MN, USA
cBayless Cancer Institute, St. Joseph’s Children’s Hospital, Tampa, FL 33607, USA
dMasonic Cancer Center, University of Minnesota, MN 55455, USA
eDepartment of Population and Public Health Sciences, University of Southern California, Los Angeles, CA, USA
fChildren’s Oncology Group, Monrovia, CA, USA
gCorresponding Author: Diana W. Lone, Bayless Cancer Institute, St. Joseph’s Children’s Hospital, Baycare Medical Group, Tampa, FL 33607, USA
Manuscript submitted May 3, 2022, accepted June 17, 2022, published online June 27, 2022
Short title: GH Deficiency in Childhood iGCT Survivors
doi: https://doi.org/10.14740/jem807
| Abstract | ▴Top |
Background: Intracranial germ cell tumor (iGCT) survivors have multiple risk factors for growth hormone (GH) deficiency, a commonly reported late effect in childhood cancer survivors. The objective of this study was to examine the prevalence of GH deficiency among childhood iGCT survivors.
Methods: Participants were previously enrolled in the Germ Cell Tumor Epidemiology Study (GaMETES), a case-parent triad study conducted using the Children’s Oncology Group registry protocols, including 216 cases with iGCTs. Data on late effects and outcomes are available for 129 iGCT cases who consented for a follow-up study including a self-administered questionnaire and medical record retrieval. GH deficiency was identified via self-report and validated through medical record review. Chi-squared and Fisher’s exact tests were used to examine cases with GH deficiency predating iGCT detection. Logistic regression was used to identify predictors of GH deficiency as a late effect.
Results: Of 129 iGCT cases who participated in the late effects study, 45% had GH deficiency, 18% had GH deficiency predating the iGCT, and 27% developed GH deficiency within a median of 19 months after diagnosis. Younger age at diagnosis, suprasellar location, and higher radiation doses were associated with GH deficiency as a late effect.
Conclusions: GH deficiency is highly prevalent as an early clinical sign for iGCT and frequently arises as an early late effect after treatment. Additional investigation is needed to address earlier detection and treatment for this highly prevalent late effect in iGCT survivors.
Keywords: Intracranial germ cell tumor; Growth hormone deficiency; Late effects
| Introduction | ▴Top |
Intracranial germ cell tumors (iGCTs) are a rare malignancy with an estimated incidence of two cases per 1,000,000 person-years in the United States [1]. Approximately 75% of iGCTs are histologically classified as germinomas, with the remainder classified as non-germinomatous germ cell tumors (NGGCTs). They have a bimodal age distribution with peaks in early childhood and in adolescence [2]. About one-quarter of iGCTs arise from the suprasellar location where the pituitary gland resides [3-5]. Contemporary treatment consists of some combination of surgery, cranial radiation and/or neoadjuvant chemotherapy resulting in 10-year overall survival rates of 90% for germinomas and 86% for NGGCTs [6].
Longitudinal studies of childhood cancer survivors have demonstrated that hypothalamic-pituitary (HP) deficiencies are among the most common late effects manifesting within 5 years after treatment exposure [7, 8]. The most common of these is growth hormone (GH) deficiency, affecting up to 46.5% of survivors treated with cranial radiation [8-11]. Risk factors for treatment-related GH deficiency include younger age at exposure, suprasellar tumor location, and a strong dose-dependent relationship with cranial and craniospinal irradiation (CSI) [4, 8-14]. These are all exposures affecting iGCT survivors placing them at higher risk for developing these deficiencies than other types of brain tumors, only behind craniopharyngiomas, medulloblastomas and low-grade gliomas [7, 15].
It is hard to know the true prevalence of GH deficiency among iGCT survivors as late effects in this group are poorly studied. iGCTs are more often studied amongst childhood brain tumors; however, even in these groups, they are considered rare and often lumped in the “other” pathology categories [8]. iGCTs histologically fall under the umbrella of GCTs, a rare and heterogenous group. GCTs were excluded from the largest long-term follow-up study, the Childhood Cancer Survivorship Study (CCSS), which is the database from which much of the late effects literature is derived. However, with improved treatments, survival outcomes approach 90% for GCTs making them account for the largest group of childhood cancer survivors living beyond 20 years from diagnosis in the United States [15]. Therefore, there is an important need to better understand late effects affecting this group of survivors, which is especially ill-defined amongst iGCTs.
GH deficiency is one of the most reported late effects and has lifelong consequences. GH deficiency left untreated can cause growth delay in children, metabolic issues in adulthood, and negatively impact quality of life [15-18]. Treatment-related GH deficits have been evaluated among iGCT survivors who were treated in adolescence or young adulthood but have not been described in younger children [4]. With a growing population of children surviving after treatment for iGCTs, there is an urgent need to better understand treatment-related toxicities that can have lasting sequalae on this group of long-term childhood cancer survivors. Therefore, the primary objective of this study was to determine the prevalence of GH deficiency, the most common HP deficit, in childhood and adolescent iGCT survivors. We hypothesized that there is a high prevalence of GH deficiency as a late effect of treatment among childhood and adolescent iGCT survivors.
| Materials and Methods | ▴Top |
Study population
Participants were recruited to this cohort of iGCT survivors from the Germ Cell Tumor Epidemiology Study (GaMETES), which is a case-parent triad study conducted using the Children’s Oncology Group (COG) Childhood Cancer Research Network (CCRN) [19]. A full description of the GaMETES study has been previously published [20]. Briefly, 866 individuals with a GCT diagnosed in any anatomic location at age of 0 - 19 years and between 2008 and 2015 were enrolled in the study. Two-hundred and sixteen had intracranial tumors. Most participants in the GaMETES (95%) consented to be contacted for future research studies.
This study consists of two parts. The first is to establish a long-term follow-up study for survivors of any type of childhood GCT. The purpose is to create a registry of data regarding late effects experienced by survivors of any type of childhood GCT to allow researchers to conduct other studies. To date, 369 individuals from the GaMETES cohort have been consented to participate in the long-term follow-up study.
The second part is to evaluate the prevalence of GH deficiency among these survivors, a pilot study utilizing this database. This will be the focus of this report. Because GH deficiency was observed almost exclusively in participants with intracranial tumors, this interim analysis was restricted to individuals with iGCT (n = 129).
This study was approved by the Institutional Review Board at the University of Minnesota. This study was conducted in compliance with the ethical standards of the responsible institution on human subjects as well as with the Helsinki Declaration. Written informed consent was obtained for all study participants, including parental consent for participants under age 18 years and self-consent for participants 18 years and older. Assent was also obtained for participants aged 8 - 17 years.
Questionnaire
Following consent, all participants over the age of 18 years or their parents were provided with a secure link to a REDCap survey. Materials developed for the CCSS were adapted and used to collect information on current health status, medication use, physical measurements, health behaviors, quality of life and school/employment history [21, 22]. For cases who are under 18 years of age at contact, we asked the parent to complete the questionnaire and cases who have reached the age of 18 completed a self-administered version. The research team periodically sent follow-up reminders to the participants to encourage them to complete the questionnaire.
Medical record review
As part of the consent process, we obtained Health Insurance Portability and Accountability Act (HIPAA) authorization from participants to collect treatment records and updated health history information from the individual COG institutions as well as from other treating institutions reported in the questionnaire. At the time of enrollment, cases are asked to name all treating hospitals and clinics. Following standard practices authorizing the release of medical information, site-specific authorizations to obtain these records are prepared and then faxed to these facilities by study staff. Medical records were requested on a rolling basis starting with iGCTs and followed by the extracranial GCTs. For this project, medical records were reviewed for tumor characteristics, treatment exposures, and hormone deficiencies. Oncology, survivorship clinic, and endocrinology clinical notes as well as lab results and imaging reports were reviewed. Data were abstracted from the medical records by two individuals (DL and CW). Discrepant data were reviewed by the primary reviewer (DL) and records were reviewed again to complete missing data. Data from medical records were used to validate self-reported questionnaire responses. There were no contradicting responses between these two sources. Where an answer was missing from one of these sources but present from the other, the available response was selected.
Variables
The primary outcome of interest for this analysis was GH deficiency. GH status was abstracted from the medical records and validated against questionnaire responses. GH status was determined by a clinician’s documented interpretation of insulin-like growth factor 1 (IGF-1), GH stimulation tests, and/or growth pattern. GH deficiency was defined by documentation of the corresponding International Classification of Disease (ICD)-9 or ICD-10 diagnosis code in the medical record. Intact GH status was defined by the presence of the above endocrinology workup but the lack of the ICD-9 or ICD-10 diagnosis code for GH deficiency. GH status was categorized as “unknown” for individuals for which no GH evaluation could be uncovered. Individuals self-reported GH status as “yes” - GH deficient, “no” - GH intact, or “not sure” in the questionnaire. In comparing GH status as determined by the medical records to self-report, there were no contradicting discrepancies (i.e., medical record documentation of GH deficiency but self-report of intact GH or vice versa). In the cases where the individual self-reported “not sure” but the medical record indicated GH deficiency, GH status was determined to be deficient. GH deficiency was further divided into those with GH deficiency prior to iGCT diagnosis and those who developed GH deficiency after iGCT diagnosis. The date of the iGCT diagnosis was retrieved from the CCRN database. The date of GH deficiency was collected where available to determine if the GH deficiency existed before or after iGCT diagnosis.
Risk factors included patient and tumor characteristics as well as treatment exposures. Patient characteristics, including age at diagnosis (0 - 14 compared to 15 - 19 years), sex, and race/ethnicity were available for all participants. Age group cutoffs were selected based on standards in other studies comparing childhood with adolescent cancers [4]. Tumor characteristics (tumor location, histology, and presence of metastatic disease) were available from pathology reports provided for all individuals as part of the CCRN protocol. Treatment data, including surgical interventions, chemotherapy protocols, radiation treatments, stem cell transplant, and salvage therapy regimens were obtained from medical records.
Tumor histology was categorized as either germinoma or NGGCT following the standard groupings used to determine treatment [5]. Primary intracranial tumor site was divided into suprasellar region (suprasellar or bifocal) versus other (pineal, thalamic, spinal, other) since the hypothalamus, pituitary stalk and pituitary gland that regulate GH production reside within the suprasellar space. Cumulative radiation doses were grouped into low (< 38 Gy), medium (38 - 51 Gy), and high (51+ Gy) dose categories. The cutoffs for each category were determined using target radiation doses per COG response-stratified guidelines [23, 24].
Other secondary anterior pituitary hormone deficiencies, including hypothyroidism, adrenal insufficiency, and hypogonadotropic hypogonadism were also explored. Dates of diagnosis for each condition were abstracted where available. Each pituitary hormone deficiency was determined based on documentation of the corresponding ICD-9 or ICD-10 diagnosis code. Each intact pituitary hormone status was determined by documented clinician interpretation of serum test results (cortisol level, thyroid-stimulating hormone and free thyroxine, follicular-stimulating hormone and luteinizing hormone). This information was unavailable for some individuals. Self-reported questionnaire responses were utilized to validate these results. Limited information about diabetes insipidus (DI), a posterior pituitary hormone deficiency, was abstracted during data collection as DI was not a specific outcome of interest since it is not considered a late effect.
Statistical analysis
Statistical analysis was performed using SAS® OnDemand for Academics: Studio software (Cary, NC). Chi-squared tests were used to test whether patient, tumor, or treatment exposure variables were associated with GH deficiency. We conducted separate analyses for patients with GH deficiency predating iGCT diagnosis and for patients with GH deficiency after iGCT diagnosis. Both groups were compared to those with intact GH regulation. Fisher’s exact test was used instead of the Chi-squared test in small sample settings (where the expected number per cell is < 5). Due to the exploratory nature of this work, multiple testing was not adjusted for. To further evaluate cases with GH deficiency after iGCT diagnosis, we used logistic regression. Estimated crude and adjusted odds ratios (ORs) are reported with 95% confidence intervals (CIs) and two-sided P-values. Predictors selected for inclusion in the final adjusted model were based on statistically significant P-values from the contingency table analysis or previous associations reported in the literature. These included age, primary tumor site, and radiation dose. Surgical intervention was found to be associated with GH deficiency by Chi-squared analysis. However, surgery has not been reported as an association in the literature and all but two cases in our study had undergone surgery making the model unstable; thus, surgery was omitted as a variable in the final model. In some studies, sex had been shown to be associated to anterior pituitary deficiency [8, 10], but in our Chi-squared and simple logistic regression analysis, no association was identified; therefore, it was not included in the final model. Crude and adjusted ORs were not calculated for groups with fewer than five participants.
| Results | ▴Top |
Characteristics of the study population
In total, this analysis includes 129 iGCT survivors with either questionnaire data (n = 114) and/or medical record data (n = 95; Fig. 1). Patient, tumor, and treatment characteristics for the 129 iGCT participants are shown in Table 1. Eighty-five (65.9%) were diagnosed with an iGCT between 0 and 14 years and 44 (34.1%) were diagnosed between 15 and 19 years. There was a male predominance (n = 92, 71.3%) and the majority were non-Hispanic white (n = 92, 71.3%). Eighty-two (68.3%) of these tumors were germinomas and 38 (31.7%) were NGGCTs. Thirty-nine (41.9%) tumors were from the suprasellar region (suprasellar or bifocal). Nearly all participants underwent a surgery but of variable extent; some received biopsies only, others subtotal resection, and some gross total resections. Seventy-three (81.1%) participants received upfront chemotherapy. Most participants with germinomas were treated with 2 - 4 cycles of neoadjuvant chemotherapy with a platinum agent and etoposide, and those with NGGCTs were treated with six cycles of ifosfamide/etoposide alternating with cycles of a platinum and etoposide. All but five received radiation therapy consisting of whole ventricular radiation with a boost to the primary site with or without CSI. Thirty-one participants received low doses of cumulative radiation, 16 received medium, and 20 received high doses.
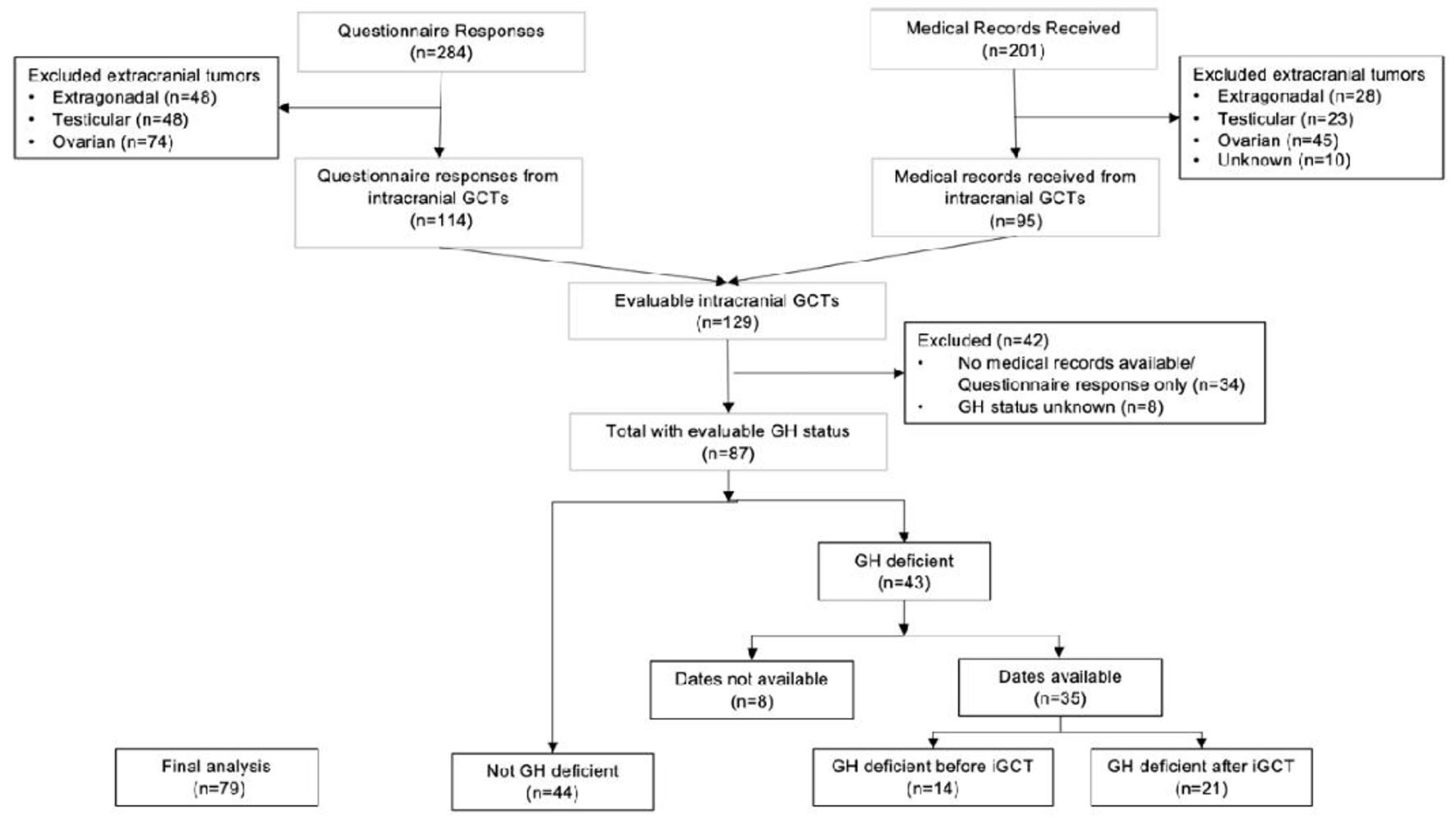 Click for large image | Figure 1. CONSORT diagram of study population. Flow chart depicting study participant inclusion. |
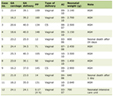 Click to view | Table 1. Characteristics of the iGCT Study Population |
GH deficiency in iGCT survivors
We included 87 cases with adequate information to determine GH status for this analysis (indeterminate for n = 8). Of these, 43 (49.4%) had GH deficiency and 44 (50.6%) did not. Of the 43 cases with GH deficiency, dates for GH diagnosis were available for 35. GH deficiency predated iGCT diagnosis in 14 of 35 (40.0%) and 21 (60.0%) developed GH deficiency after iGCT diagnosis (Fig. 1). Analysis was done separately for those with GH deficiency before (n = 14) and after iGCT diagnosis (n = 21) compared to those with intact GH (n = 44).
GH deficiency predating iGCT diagnosis
We found that 14 out of 79 (17.7%) of the iGCT survivors had GH deficiency prior to detection of the tumor. This was more common among younger children who were diagnosed with an iGCT between 0 and 14 years (12.7%) compared to those diagnosed in adolescence (5.1%). This was also more common among individuals who would subsequently develop tumors in the suprasellar region compared to tumors in other intracranial sites (P ≤ 0.0001, Table 2). GH deficiency was diagnosed within a median of 4.5 months (range 0 - 28 months) prior to detection of the iGCT. Four of these individuals were started on GH replacement therapy prior to detection of the iGCT. All of them discontinued GH therapy upon detection of the tumor.
Click to view | Table 2. Characteristics of Cases With GH Deficiency Before and After iGCT Diagnosis |
GH deficiency after iGCT diagnosis
Twenty-one of the 79 (26.6%) evaluable iGCT survivors did not have GH deficiency prior to detection of the iGCT but developed GH deficiency sometime later. This was more common among children diagnosed with an iGCT between 0 and 14 years (20.3%) than in adolescents (6.3%). In the adjusted analysis, suprasellar tumor location (OR: 38.7, 95% CI: 3.43 - 436.21) and age at diagnosis younger than 15 years (OR: 4.87, 95% CI: 0.741 - 32.04) were found to be associated with GH deficiency (Table 3). The odds of GH deficiency were estimated to be higher among those treated with medium doses of radiation compared to those treated with low dose (OR: 10.68, 95% CI: 0.93 - 122.86), and for high compared to low dose (OR: 9.03, 95% CI: 0.93 - 122.86), but these estimates were imprecise. The observed median time from iGCT diagnosis to GH deficiency detection was 19.0 months (range 4 - 82 months). Seventeen cases were treated with GH replacement that was initiated within a median of 2.0 months after diagnosis of GH deficiency. None of the participants treated with GH replacement developed a secondary malignancy at the time of this evaluation (median follow-up of 6.7 years). One had recurrence of the primary NGGCT after initiation of GH replacement. This individual was started on GH therapy 29 months after the primary NGGCT, and later developed recurrent disease after another 50 months. GH therapy was subsequently discontinued, and this individual underwent additional courses of salvage therapy for multiply recurrent disease.
Click to view | Table 3. Predictors of GH Deficiency After iGCT Diagnosis |
Other pituitary hormone deficiencies
Of 95 cases, 45 (47.3%) iGCT participants had hypothyroidism. Dates for detection for hypothyroidism were available for 33 of 45 participants. Of these 33 cases, 17 (51.5%) had hypothyroidism predating the iGCT diagnosis, and 16 (48.5%) developed hypothyroidism after the iGCT diagnosis. Thirty-five out of the 95 cases (36.8%) had adrenal insufficiency and dates were available for 29. Twenty-five of 29 (86.2%) were detected prior to the iGCT diagnosis and four (13.8%) were detected afterwards. Thirty-three of 95 (34.7%) had hypogonadism, and dates were available for 24, including 12 (50.0%) predating the iGCT and 12 (50.0%) arising after the iGCT diagnosis (Fig. 2).
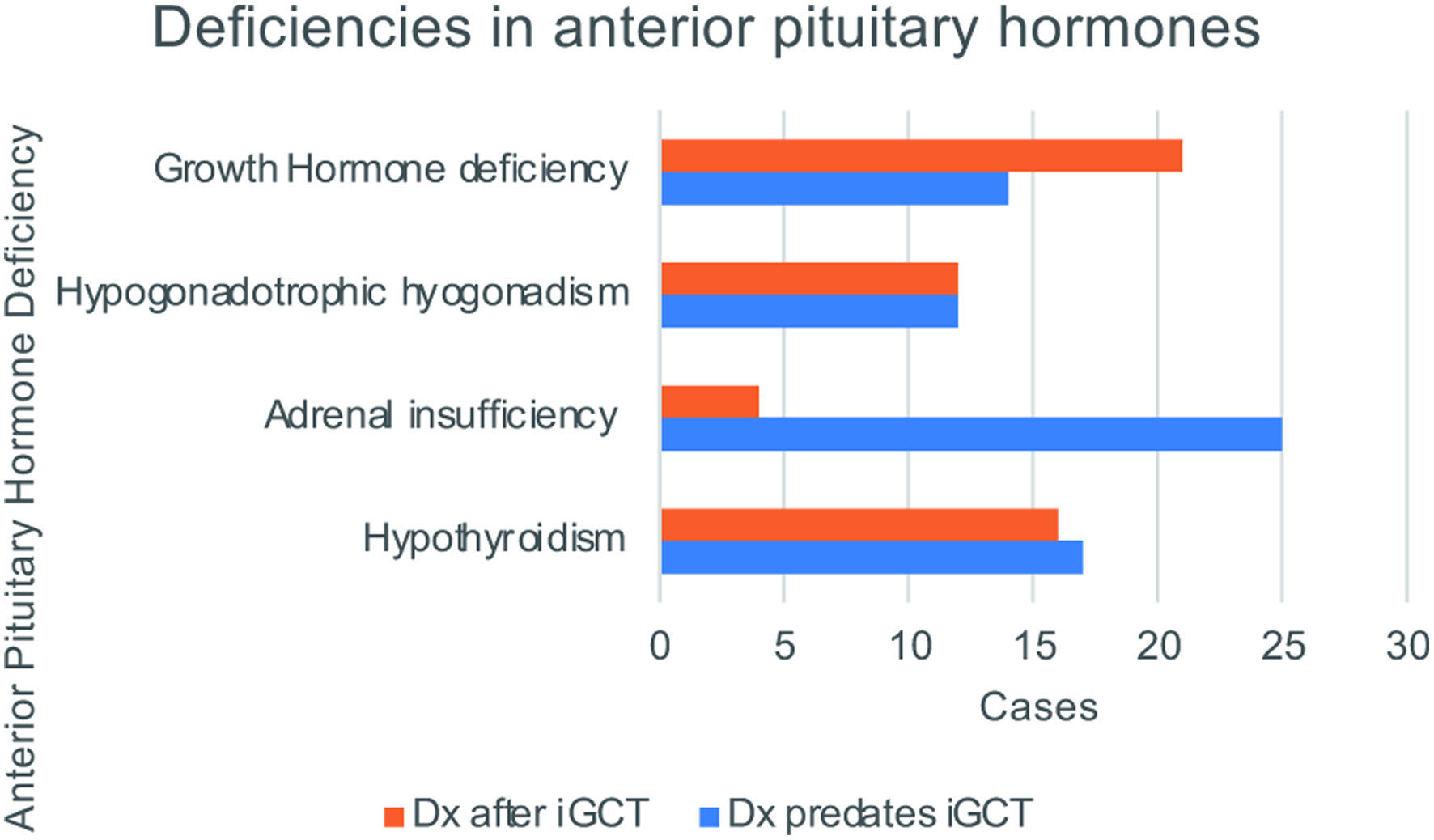 Click for large image | Figure 2. Deficiencies in anterior pituitary hormones. Of the full cohort of evaluable iGCT survivors, the total number of cases with each anterior pituitary hormone deficiency that predated the iGCT diagnosis is shown in blue, and the total cases with each respective hormone deficiency detected after iGCT diagnosis is shown in orange. |
DI (vasopressin deficiency) was not specifically investigated in this study as it is not considered a late effect, but at least 15 cases had DI preceding iGCT diagnosis, six of whom had both DI and GH deficiency and nine with DI but no GH deficiency before tumor diagnosis.
| Discussion | ▴Top |
The overall prevalence of GH deficiency in our study of iGCT survivors was 45.3% (43 of 95). This approximates the estimated point prevalence for GH deficiency reported as 47.5% among adult survivors of childhood cancers treated with cranial radiation > 18 Gy in the St. Jude LIFE cohort [10]. Interestingly, a large percentage of the iGCT survivors with GH deficiency were diagnosed prior to tumor detection (17.7%), suggesting that GH deficiency is an important problem both before and after iGCT diagnosis. Therefore, it is essential to consider GH deficiency as an early clinical manifestation of an iGCT separately from GH deficiency as a late effect of cancer therapy.
We suspect this is an underestimation of the true prevalence of GH deficiency, both before and after the iGCT diagnosis as not all individuals received a GH evaluation. Few individuals were tested for GH deficiency prior to iGCT diagnosis as growth deceleration was infrequently the presenting symptom. Children who were pre-pubertal following completion of treatment were more likely to be evaluated for GH deficiency than adolescents who had already achieved their adult height. However, it should be noted that growth deceleration is not necessary nor sufficient to diagnose GH deficiency and individuals who have reached their adult height can still be GH deficient and can still benefit from GH replacement.
We observed a strong association between the suprasellar tumor site and early manifestation of GH deficiency. Our finding aligns with many studies which have reported endocrinopathies as a common presenting manifestation for suprasellar intracranial tumors [25, 26]. GH deficiency predating iGCT diagnosis was more common among children diagnosed with iGCT at 0 - 14 years (71.4%) than 15 - 19 years (28.6%). This highlights the importance of a thorough evaluation for a brain tumor in children who present with growth failure and GH deficiency. A previous study showed that growth deceleration predated radiologic iGCT detection by as long as 1.1 years, though this study included only 12 iGCTs [27]. In our study, GH deficiency was detected within a median of 4.5 months (range 0 - 28 months) prior to iGCT diagnosis. This suggests that ongoing surveillance for a brain tumor in children with GH deficiency may be needed for upwards of 2 years. Although brain imaging has a low yield in asymptomatic short children, the yield of identifying structural abnormalities or tumors with brain imaging is increased in children with growth deceleration and GH deficiency [28]. There were four individuals within our study population who initially had normal brain magnetic resonance images when evaluated for HP deficiencies but later developed radiographically detectable iGCTs.
While GH deficiency can sometimes be a presenting sign for an intracranial neoplasm, other endocrinopathies are more common presenting symptoms of intracranial neoplasm, especially DI [26, 27]. Fifty-one percent of those with hypothyroidism, 86.2% of those with adrenal insufficiency, and 50.0 % of those with hypogonadotropic hypogonadism had detectable hormone deficiencies predating their iGCT diagnosis. This highlights the need for ongoing close follow-up with attention to malignancy in children with endocrinopathies even if there was no evidence for a tumor at the time of the initial evaluation. We recommend a low threshold for obtaining cranial imaging to assess for an intracranial tumor in children with HP deficiencies without another etiology and a consideration for repeated imaging within up to 2 years from diagnosis of an HP deficiency, especially if there is a clinical change or progression to multiple HP deficiencies. Since this was a retrospective cohort study, we were unable to determine if any iGCT diagnoses were missed or delayed in individuals who presented with HP deficiencies predating the tumor. This should be evaluated in future studies.
Our prevalence rate for GH deficiency as a late effect was 26.6% (21 out of the evaluable 79). This is higher than the 12.5% reported among childhood brain tumor survivors who were treated with > 30 Gy radiation [8]. These differences may be affected by selection bias if those who experienced late effects were more compelled to complete the questionnaire. This might also be attributed to a different distribution of radiation treatment doses that were used for the heterogenous group of brain tumors in that study compared to the more uniform treatment received by participants in our study. We also report a higher prevalence (16.7%) for GH deficiency as a late effect in adolescent young adults (AYAs) with iGCT than has been reported in another study (1%) [4]. We additionally found that suprasellar tumor location was very strongly associated with GH deficiency after adjusting for radiation dose and age at diagnosis. This may be attributed to the local destruction of the pituitary gland by the tumor itself compounded by radiation targeted at this site. Our findings are consistent with other studies that have shown that younger age at diagnosis and suprasellar tumor location are the major risk factors for GH deficiency as a late effect [8-10, 12, 29, 30].
The dose-dependent effect of cranial radiation on GH deficiency has been repeatedly demonstrated in the literature even for patients who received as little as 18 Gy of craniospinal radiation [10, 31] and risk increases in a dose-dependent manner [4, 9, 11-14, 29]. However, variable dose cutoffs have been used to define low versus high doses of radiation. In this study, the radiation dosing categories were created using target radiation doses per COG guidelines. According to the most common treatment regimens during the study period, radiation doses were selected based on tumor response to neoadjuvant chemotherapy and the use of CSI was minimized. Therefore, germinoma treatment included either 30, 36, or 45 Gy of cranial radiation (per ACNS0232 and ACNS1123) [23] and NGGCT treatment consisted of either 45 or 54 Gy of cranial radiation (per ACNS0122 and ACNS1123) [24]. The dosing ranges we used to categorize radiation exposure were expanded to capture those who received slightly above or below those targets. A cumulative dose of < 38 Gy captures those receiving a target dose of 30 or 36 Gy and were categorized as low dose. Those treated with a target of 45 Gy were grouped into the medium dose group (38 - 51 Gy), and those treated with a goal of 54 Gy were categorized as receiving high dose (51 Gy +). We observed a similar increase in risk for patients in the medium and high radiation dose categories, although we had limited power to detect differences between these groups due to small sample size.
Like other studies of childhood brain tumor survivors who were treated with cranial radiation, the median time from cancer to GH deficiency as a late effect was short [7, 8]. In our study, the median time from iGCT diagnosis to GH deficiency detection was only 19 months. However, this timeframe may be an over-estimate as it is dependent upon when providers initiated the evaluation for GH deficiency. Because the general practice is to delay GH replacement therapy until a patient is at least 12 months past completion of cancer-directed therapy to avoid GH therapy during the most likely period of tumor recurrence [32, 33], many of our participants were not evaluated for GH deficiency until they were nearly 12 months post cancer treatment. Those who were evaluated prior to 12 months off-therapy delayed initiation of GH replacement until that point. The median time from diagnosis of GH deficiency until treatment initiation was short at about 2 months.
The current expert recommendation for when it is safe to initiate GH therapy remains controversial; however, the association between GH therapy and secondary malignancies has not been demonstrated in more recent studies of childhood cancer survivors [15, 34, 35]. We further contribute to this evidence as none of the participants treated with GH replacement in our study developed a secondary malignancy within our median follow-up period of 6.7 years, and only one patient had recurrence of the primary tumor. Further studies are needed to define the relationship between GH replacement agents and the risk for secondary malignancies as childhood cancer survivors could benefit from earlier evaluation for GH deficiency and earlier initiation of GH replacement.
Other pituitary hormone deficiencies are also important late effects. However, GH deficiency was more prevalent as a late effect than as a condition predating the iGCT diagnosis. Hypothyroidism, adrenal insufficiency, and hypogonadism more commonly preceded the iGCT diagnosis. In many cases, multiple of these pituitary deficiencies were present preceding the iGCT diagnosis. This suggests that panhypopituitarism is very common as both an early clinical presentation and as a late effect of iGCT therapy.
A strength of this analysis is the adequate sample size of children and AYAs with iGCTs. To our knowledge, this is the largest described cohort of childhood iGCTs. One hundred and twenty-nine of the 216 (59.7%) iGCT survivors from the broader GaMETES study are enrolled in the long-term follow-up study. There was little demographic variability in the iGCT sample compared to the iGCTs within the original GaMETES cohort in terms of age at diagnosis, sex, and histology, suggesting that this sample is representative of the larger cohort for the outcomes of interest. Hispanic participants were under-represented within our sample; 12.5% within the GaMETES cohort were Hispanic compared to 7.8% within our study. Enrollment for Spanish-speaking participants did occur later in our consent process due to delay in approval of our Spanish-translated consent forms and questionnaire. Additionally, language barrier, despite having Spanish-translated materials might contribute to lower enrollment rates. However, there is no significant difference in incidence rates for iGCTs among Hispanic and non-Hispanic individuals [2], so this difference in distribution is not expected to affect the validity of our outcomes.
Findings from this study confirm previously published literature but expand our knowledge regarding late effects experienced by children and AYAs who were treated for iGCTs. This fills an important gap in the literature because this has not previously been studied within childhood iGCT survivors. Cases in this study were treated with contemporary treatment regimens making these late effects highly relevant to current survivorship care.
There are also several limitations to this study. We did not have sufficient data available to detect the impact of varying modalities of radiation delivery. In addition, our sample size for those who developed GH deficiency as a late effect was small after excluding those with insufficient data on GH status and those who developed GH deficiency predating cancer. Dates for end of radiation therapy were missing for a subset of the participants, restricting our ability to attribute GH deficiency to radiation rather than to all cancer-directed therapies. We did not have sufficient data to determine the prevalence of the other pituitary hormone deficiencies. Lastly, our analysis is restricted by the short follow-up time of 6.7 years. While this is an appropriate length of time to assess GH deficiency, which typically appears in the early post treatment period, it is insufficient for detecting secondary malignancies. It should be noted that this work was exploratory and meant to identify potential associations between iGCT diagnosis and GH deficiency.
In conclusion, 45.3% of evaluable iGCT survivors in this study had GH deficiency, including 17.7% with GH deficiency that predated their iGCT diagnosis. Individuals with tumors in the suprasellar region had the highest prevalence of pre-diagnosis GH deficiency, suggesting that local damage to the hypothalamus, pituitary stalk and pituitary gland by the tumor is the main source for GH deficiency in this group. Among the 26.6% of individuals who developed GH deficiency after iGCT diagnosis, younger age at iGCT diagnosis, suprasellar tumor location, and radiation dose were strongly associated with GH deficiency. The median time to detection of GH deficiency was 19 months after cancer diagnosis. Additional investigation is needed to address earlier detection and treatment for this highly prevalent late effect in iGCT survivors.
Acknowledgments
None to declare.
Financial Disclosures
This work was supported by the National Institutes of Health (NIH, T32 CA099936 to DL; R01 CA151284 to JNP; NCTN Operations Center Grant (U10 CA180886); and NCTN Statistics & Data Center Grant (U10CA180899), and the Children’s Cancer Research Fund, Minneapolis, MN.
Conflict of Interest
None to declare.
Informed Consent
Written informed consent was provided by parents of germ cell tumor survivors under age 18 years at enrollment. Study participants aged 18 years or older provided written informed consent. Assent was obtained from participants aged 8 - 17 years.
Author Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Diana W. Lone, Jeannette Sample, Aubrey K. Hubbard, Michelle Roesler and Jenny N. Poynter. The first draft of the manuscript was written by Diana W. Lone and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Abbreviations
AYA: adolescent young adult; CCRN: Childhood Cancer Research Network; CCSS: Childhood Cancer Survivorship Study; CI: confidence interval; COG: Children’s Oncology Group; CSI: craniospinal irradiation; GaMETES: Germ Cell Tumor Epidemiology Study; GCT: germ cell tumor; GH: growth hormone; HIPAA: Health Insurance Portability and Accountability Act; HP: hypothalamic-pituitary; ICD: International Classification of Disease; iGCT: intracranial germ cell tumor; NGGCT: non-germinomatous germ cell tumor; NIH: National Institutes of Health; OR: odds ratio
| References | ▴Top |
- Villano JL, Propp JM, Porter KR, Stewart AK, Valyi-Nagy T, Li X, Engelhard HH, et al. Malignant pineal germ-cell tumors: an analysis of cases from three tumor registries. Neuro Oncol. 2008;10(2):121-130.
doi pubmed - Poynter JN, Fonstad R, Tolar J, Spector LG, Ross JA. Incidence of intracranial germ cell tumors by race in the United States, 1992-2010. J Neurooncol. 2014;120(2):381-388.
doi pubmed - Echevarria ME, Fangusaro J, Goldman S. Pediatric central nervous system germ cell tumors: a review. Oncologist. 2008;13(6):690-699.
doi pubmed - Wong J, Goddard K, Laperriere N, Dang J, Bouffet E, Bartels U, Hodgson D, et al. Long term toxicity of intracranial germ cell tumor treatment in adolescents and young adults. J Neurooncol. 2020;149(3):523-532.
doi pubmed - Goldman S, Bouffet E, Fisher PG, Allen JC, Robertson PL, Chuba PJ, Donahue B, et al. Phase II trial assessing the ability of neoadjuvant chemotherapy with or without second-look surgery to eliminate measurable disease for nongerminomatous germ cell tumors: a children's oncology group study. J Clin Oncol. 2015;33(22):2464-2471.
doi pubmed - Lo AC, Hodgson D, Dang J, Tyldesley S, Bouffet E, Bartels U, Cheng S, et al. Intracranial germ cell tumors in adolescents and young adults: a 40-year multi-institutional review of outcomes. Int J Radiat Oncol Biol Phys. 2020;106(2):269-278.
doi pubmed - Lawson SA, Horne VE, Golekoh MC, Hornung L, Burns KC, Fouladi M, Rose SR. Hypothalamic-pituitary function following childhood brain tumors: analysis of prospective annual endocrine screening. Pediatr Blood Cancer. 2019;66(5):e27631.
doi pubmed - Clement SC, Schouten-van Meeteren AY, Boot AM, Claahsen-van der Grinten HL, Granzen B, Sen Han K, Janssens GO, et al. Prevalence and risk factors of early endocrine disorders in childhood brain tumor survivors: a nationwide, multicenter study. J Clin Oncol. 2016;34(36):4362-4370.
doi pubmed - Chemaitilly W, Cohen LE, Mostoufi-Moab S, Patterson BC, Simmons JH, Meacham LR, van Santen HM, et al. Endocrine late effects in childhood cancer survivors. J Clin Oncol. 2018;36(21):2153-2159.
doi pubmed - Chemaitilly W, Li Z, Huang S, Ness KK, Clark KL, Green DM, Barnes N, et al. Anterior hypopituitarism in adult survivors of childhood cancers treated with cranial radiotherapy: a report from the St Jude Lifetime Cohort study. J Clin Oncol. 2015;33(5):492-500.
doi pubmed - Gleeson HK, Shalet SM. The impact of cancer therapy on the endocrine system in survivors of childhood brain tumours. Endocr Relat Cancer. 2004;11(4):589-602.
doi pubmed - Darzy KH, Shalet SM. Hypopituitarism as a consequence of brain tumours and radiotherapy. Pituitary. 2005;8(3-4):203-211.
doi pubmed - Shalet SM, Beardwell CG, Pearson D, Jones PH. The effect of varying doses of cerebral irradiation on growth hormone production in childhood. Clin Endocrinol (Oxf). 1976;5(3):287-290.
doi pubmed - Rappaport R, Brauner R. Growth and endocrine disorders secondary to cranial irradiation. Pediatr Res. 1989;25(6):561-567.
doi pubmed - Pekic S, Stojanovic M, Popovic V. Controversies in the risk of neoplasia in GH deficiency. Best Pract Res Clin Endocrinol Metab. 2017;31(1):35-47.
doi pubmed - Molitch ME, Clemmons DR, Malozowski S, Merriam GR, Vance ML, Endocrine S. Evaluation and treatment of adult growth hormone deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011;96(6):1587-1609.
doi pubmed - Follin C, Thilen U, Osterberg K, Bjork J, Erfurth EM. Cardiovascular risk, cardiac function, physical activity, and quality of life with and without long-term growth hormone therapy in adult survivors of childhood acute lymphoblastic leukemia. J Clin Endocrinol Metab. 2010;95(8):3726-3735.
doi pubmed - Mukherjee A, Tolhurst-Cleaver S, Ryder WD, Smethurst L, Shalet SM. The characteristics of quality of life impairment in adult growth hormone (GH)-deficient survivors of cancer and their response to GH replacement therapy. J Clin Endocrinol Metab. 2005;90(3):1542-1549.
doi pubmed - Musselman JR, Spector LG, Krailo MD, Reaman GH, Linabery AM, Poynter JN, Stork SK, et al. The Children's Oncology Group Childhood Cancer Research Network (CCRN): case catchment in the United States. Cancer. 2014;120(19):3007-3015.
doi pubmed - Poynter JN, Richardson M, Roesler M, Krailo M, Amatruda JF, Frazier AL. Family history of cancer in children and adolescents with germ cell tumours: a report from the Children's Oncology Group. Br J Cancer. 2018;118(1):121-126.
doi pubmed - Robison LL, Armstrong GT, Boice JD, Chow EJ, Davies SM, Donaldson SS, Green DM, et al. The childhood cancer survivor study: a National Cancer Institute-supported resource for outcome and intervention research. J Clin Oncol. 2009;27(14):2308-2318.
doi pubmed - Leisenring WM, Mertens AC, Armstrong GT, Stovall MA, Neglia JP, Lanctot JQ, Boice JD, Jr., et al. Pediatric cancer survivorship research: experience of the Childhood Cancer Survivor Study. J Clin Oncol. 2009;27(14):2319-2327.
doi pubmed - Khatua S, Fangusaro J, Dhall G, Boyett J, Wu S, Bartels U. GC-17 the children's oncology group (COG) current treatment approach for children with newly diagnosed central nervous system (CNS) localized germinoma (ACNS1123 STRATUM 2). Neuro Oncol. 2016;18(suppl 3):iii45-iii46.
doi - Fangusaro J, Wu S, MacDonald S, Murphy E, Shaw D, Bartels U, Khatua S, et al. Phase II trial of response-based radiation therapy for patients with localized CNS nongerminomatous germ cell tumors: a children's oncology group study. J Clin Oncol. 2019;37(34):3283-3290.
doi pubmed - Jorsal T, Rorth M. Intracranial germ cell tumours. A review with special reference to endocrine manifestations. Acta Oncol. 2012;51(1):3-9.
doi pubmed - Pal R, Rai A, Vaiphei K, Gangadhar P, Gupta P, Mukherjee KK, Singh P, et al. Intracranial germinoma masquerading as secondary granulomatous hypophysitis: a case report and review of literature. Neuroendocrinology. 2020;110(5):422-429.
doi pubmed - Taylor M, Couto-Silva AC, Adan L, Trivin C, Sainte-Rose C, Zerah M, Valteau-Couanet D, et al. Hypothalamic-pituitary lesions in pediatric patients: endocrine symptoms often precede neuro-ophthalmic presenting symptoms. J Pediatr. 2012;161(5):855-863.
doi pubmed - Collett-Solberg PF, Jorge AAL, Boguszewski MCS, Miller BS, Choong CSY, Cohen P, Hoffman AR, et al. Growth hormone therapy in children; research and practice - a review. Growth Horm IGF Res. 2019;44:20-32.
doi pubmed - Merchant TE, Rose SR, Bosley C, Wu S, Xiong X, Lustig RH. Growth hormone secretion after conformal radiation therapy in pediatric patients with localized brain tumors. J Clin Oncol. 2011;29(36):4776-4780.
doi pubmed - Pradhan KR, Chen Y, Moustoufi-Moab S, Krull K, Oeffinger KC, Sklar C, Armstrong GT, et al. Endocrine and metabolic disorders in survivors of childhood cancers and health-related quality of life and physical activity. J Clin Endocrinol Metab. 2019;104(11):5183-5194.
doi pubmed - Viana MB, Vilela MI. Height deficit during and many years after treatment for acute lymphoblastic leukemia in children: a review. Pediatr Blood Cancer. 2008;50(2 Suppl):509-516; discussion 517.
doi pubmed - Raman S, Grimberg A, Waguespack SG, Miller BS, Sklar CA, Meacham LR, Patterson BC. Risk of neoplasia in pediatric patients receiving growth hormone therapy—a report from the pediatric endocrine society drug and therapeutics committee. J Clin Endocrinol Metab. 2015;100(6):2192-2203.
doi pubmed - Grimberg A, DiVall SA, Polychronakos C, Allen DB, Cohen LE, Quintos JB, Rossi WC, et al. Guidelines for growth hormone and insulin-like growth factor-I treatment in children and adolescents: growth hormone deficiency, idiopathic short stature, and primary insulin-like growth factor-I deficiency. Horm Res Paediatr. 2016;86(6):361-397.
doi pubmed - Patterson BC, Chen Y, Sklar CA, Neglia J, Yasui Y, Mertens A, Armstrong GT, et al. Growth hormone exposure as a risk factor for the development of subsequent neoplasms of the central nervous system: a report from the childhood cancer survivor study. J Clin Endocrinol Metab. 2014;99(6):2030-2037.
doi pubmed - Ergun-Longmire B, Mertens AC, Mitby P, Qin J, Heller G, Shi W, Yasui Y, et al. Growth hormone treatment and risk of second neoplasms in the childhood cancer survivor. J Clin Endocrinol Metab. 2006;91(9):3494-3498.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.