

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website http://www.jofem.org |
Case Report
Volume 5, Number 1-2, April 2015, pages 184-188
Soft Tissue Metastasis of Parathyroid Carcinoma: Description of a Difficult Case
Ana Filipa Martinsa, f, Joao Martin Martinsa, Sonia do Valea, Carl Christofer Juhlinb, Lucas Batistac, Marco Francod, Dolores Lopez-Presad, Gabriel Miltenberger-Miltenyie
aEndocrine Department, North Lisbon Hospital Center and Lisbon Medical School, Lisbon, Portugal
bDepartment of Oncology and Pathology, Karolinska Institutet, Karolinska University Hospital, Stockholm, Sweden
cSurgery I Department, North Lisbon Hospital Center and Lisbon Medical School, Lisbon, Portugal
dPathology Department, North Lisbon Hospital Center and Lisbon Medical School, Lisbon, Portugal
eMolecular Medicine Institute, North Lisbon Hospital Center and Lisbon Medical School, Lisbon, Portugal
fCorresponding Author: Ana Filipa Martins, Endocrine Department, North Lisbon Medical Center, Santa Maria Hospital and Lisbon Medical School, 6th Floor, Av. Prof. Egas Moniz, 1649-028 Lisboa, Portugal
Manuscript accepted for publication October 30, 2014
Short title: Soft Tissue Metastasis of Parathyroid Cancer
doi: http://dx.doi.org/10.14740/jem240w
| Abstract | ▴Top |
Parathyroid carcinoma is an extremely rare cause of hyperparathyroidism. Histologic diagnosis may be challenging. DAB, male aged 66 was submitted to subtotal gastrectomy because of a localized gastric adenocarcinoma (intestinal type). A mild primary hyperparathyroidism was simultaneously found. Calcium oxalate nephrolithiasis was diagnosed 16 years before. Sonography revealed a nodular lesion in the right lower thyroid pole, while NMR showed a nodular lesion at the left lower thyroid pole. The sestamibi scan found a late image hyperfixation at the right lower pole. Bilateral inferior parathyroidectomy was performed and revealed on both sides adenoma and moderate nuclear polymorphism and no mitotic figures; on the left side a central cystic area was found. Clinical and biochemical cure apparently followed. Six months later a presumed unrelated nodular lesion in the right thigh muscular mass was removed and found to have numerous mitotic figures and immunoreactivity for PTH. A sestamibi scan showed marked hyperfixation in the right leg and PET-FDG-F18 hyperfixated the right quadricipital area. There is evidence for a long standing disease since nephrolithiasis was found some years before. Long standing hyperparathyroidism and increased gastric acid secretion can lead to chronic gastritis, intestinal metaplasia and gastric adenocarcinoma. There is evidence for evolution along bilateral hyperplasia, adenoma and carcinoma since bilateral disease was present and left cystic nonfunctioning lesion was found. Most surprisingly a single metastatic lesion was later found in a very unusual location and this lesion was highly undifferentiated in sharp contrast with the primitive lesion.
Keywords: Parathyroid carcinoma; Hyperparathyroidism; Metastasis
| Introduction | ▴Top |
Parathyroid carcinoma is a very unusual condition accounting for less than 0.5% of all cases of primary hyperparathyroidism, by itself a rare disease with an estimated incidence of 1:1,000 per year [1, 2]: since 1930, less than 500 cases have been reported in the English literature, although recent data suggest that it may be more common than previously considered [1, 2].
The clinical course is generally distinctive with very high levels of parathormone (PTH), serious hypercalcemia, nephrolithiasis and bone involvement, while acute pancreatitis or peptic ulcer disease may occur. Parathyroid glands are generally grossly enlarged. However an indolent course is common, local growth being generally slow and regional lymphatic (30%) or blood-borne long distance metastasis, mostly in the lung (40%) or liver (10%) being less common and later events [1, 2]. The pathological distinction between benign and malignant tumors may be difficult: only local invasion and/or distant metastasis unequivocally establish malignancy [1-3]. Surgical treatment is the only potential curative therapy. Radiotherapy and chemotherapy are uniformly ineffective, even if new calcimimetic agents may offer some help in controlling hypercalcemia [1-4].
Parathyroid carcinoma has been described in relation to multiple endocrine neoplasia (MEN) type 1 MENIN mutation (locus 11q13), type 2 RET mutations (locus 10q11.2), the hyperparathyroidism-jaw tumor (HPT-JT) syndrome and familial isolated primary hyperparathyroidism HRPT2 mutations (locus 1q21-q31) [1-4]. All of them are autosomal dominant disorders, with constitutional mutations [1, 2]. On the other hand several acquired tumoral genetic abnormalities have been described in sporadic or familial cases of parathyroid cancer including cyclin D1 (CCND1 or PRAD1) mutation (locus 11q13), mutation of the RB gene (locus 13q14), mutation of p53 (locus 17p13.1), 46,XY,t(3;4), 47,XY+7, 47XY, inv (9) as well as loss of heterozygote (LOH) at several of these loci. More generally oncogenes in 1q, 5q, 9q, 16p, 19p and Xq and tumor suppression genes in 1p, 3q, 4q, 13q and 21q may be implicated in the pathogenesis of parathyroid carcinoma. Neck irradiation is the only other known factor [1, 2].
| Case Report | ▴Top |
DAB, a Caucasian male, aged 66 years, living in Lisbon, was submitted to subtotal gastrectomy in October 2009 because of a gastric adenocarcinoma. Endoscopy revealed an ulcerated lesion 10 × 8 mm, in the gastric antrum, centered at the lesser curvature, close to the pylorus. Histological examination revealed a well differentiated tubular adenocarcinoma reaching the submucosa (intestinal type according to Lauren classification), while chronic atrophic gastritis with intestinal metaplasia was evident. Surgical resection was complete, with no evidence of epiploon or lymphatic involvement. Abdominal CT scan did not show any evidence of disseminated disease. CEA, CA19.9, CA72.4, AFP and NSE were negative. There was no family history of gastrointestinal neoplasia.
During presurgical evaluation a mild primary hyperparathyroidism was noted and after surgery the patient was referred for endocrine consultation. There was a personal history of calcium oxalate nephrolithiasis since age 50 and high blood pressure (HBP) since age 40, now using losartan 100 mg, hydrochlorothiazide 12.5 mg, furosemide 40 mg, and spironolactone 25 mg daily, with controlled blood pressure levels and no cardiovascular symptomatology. Analytical evaluation confirmed primary hyperparathyroidism: serum calcium 12.1 mg/dL (reference values (RV): 8.6 - 10.2), serum phosphates 2.2 mg/dL (RV: 2.4 - 5.1), serum magnesium 1.8 mg/dL (RV: 1.3 - 2.7), ionized serum calcium 1.67 mmol/L (RV: 1.13 - 1.32), PTH 187 pg/mL (RV: 14 - 72), 25-hydroxyvitamin D 11.2 ng/mL (RV: 9 - 56), and calcitonin: 4.7 pg/mL (RV: < 8.4). There was no osteopenia or osteoporosis on bone densitometry and osteocalcin and deoxypyridinoline were normal. Cervical sonography revealed a nodular lesion 12 × 5 mm adjacent to the lower right pole of the thyroid (Fig. 1a), while cervical NMR revealed a 22 mm nodular lesion at the lower left thyroid pole with T1 hyposignal and T2 hypersignal. Sestamibi scintigraphy showed late images in the right lower pole of the thyroid (Fig. 1b).
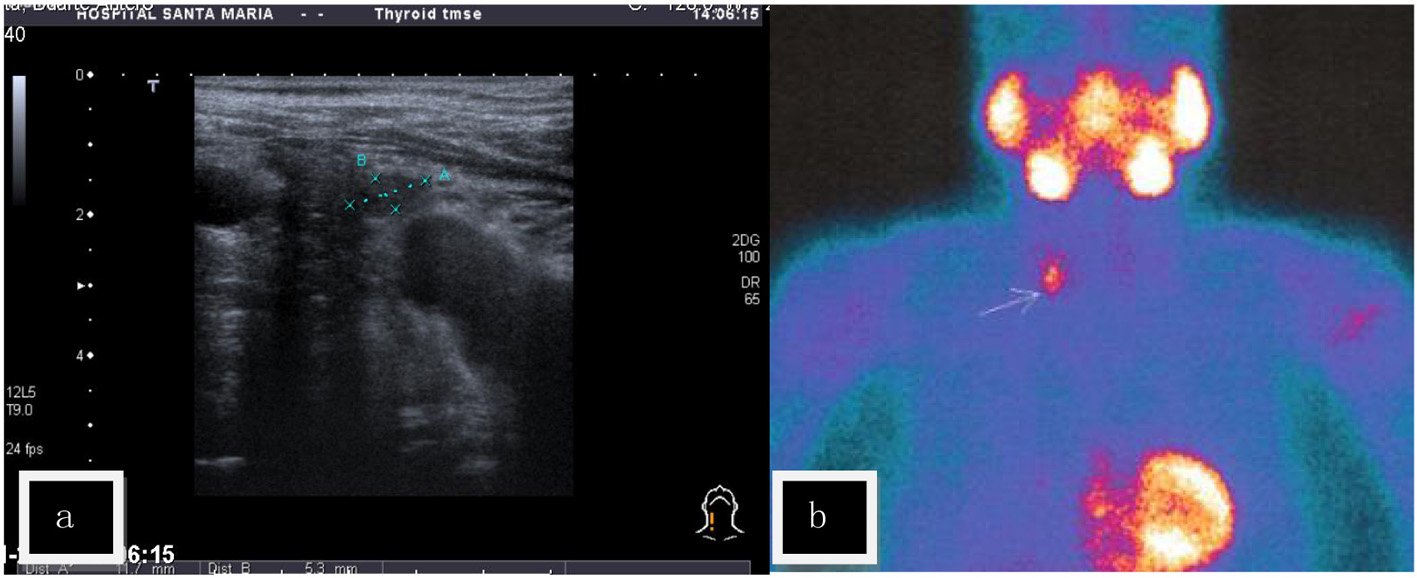 Click for large image | Figure 1. (a) Cervical sonography (US) - a nodular lesion was found contiguous to the lower right pole of the thyroid gland. (b) Sestamibi scintigraphy - positive late fixation at the right lower pole of the thyroid. |
After surgical exploration of the neck, right (1) and left (2) inferior parathyroidectomy were performed in April 2010. Pathological examination revealed: 1) parathyroid gland with 10 mm and 2.4 g; proliferation of chief cells in small nests with scarce stroma; the cells present clear cytoplasm and moderate nuclear polymorphism without mitoses; thin capsule at the borders; parafibromin negative nucleolar staining; 2) parathyroid gland with 30 × 25 mm and 7 g with a central cystic area; there is a thick capsule and thick fibrous septa dividing the gland in two nodular lesions; again proliferation of chief cells in small nests among fibrotic areas with hemosiderophages; no evidence of mitoses, neither of capsular or vascular invasion; parafibromin negative nucleolar staining with positive staining for the adenomatous polyposis coli (APC) protein (Fig. 2). In conclusion, parathyroid adenomas, with some aspects regarding the left parathyroid gland are suggestive of atypical adenoma. Negative parafibromin nucleolar staining suggests the possibility of a HRPT2 mutation and increases the likelihood of malignant potential. Postoperative evolution was uneventful with no need for further therapy.
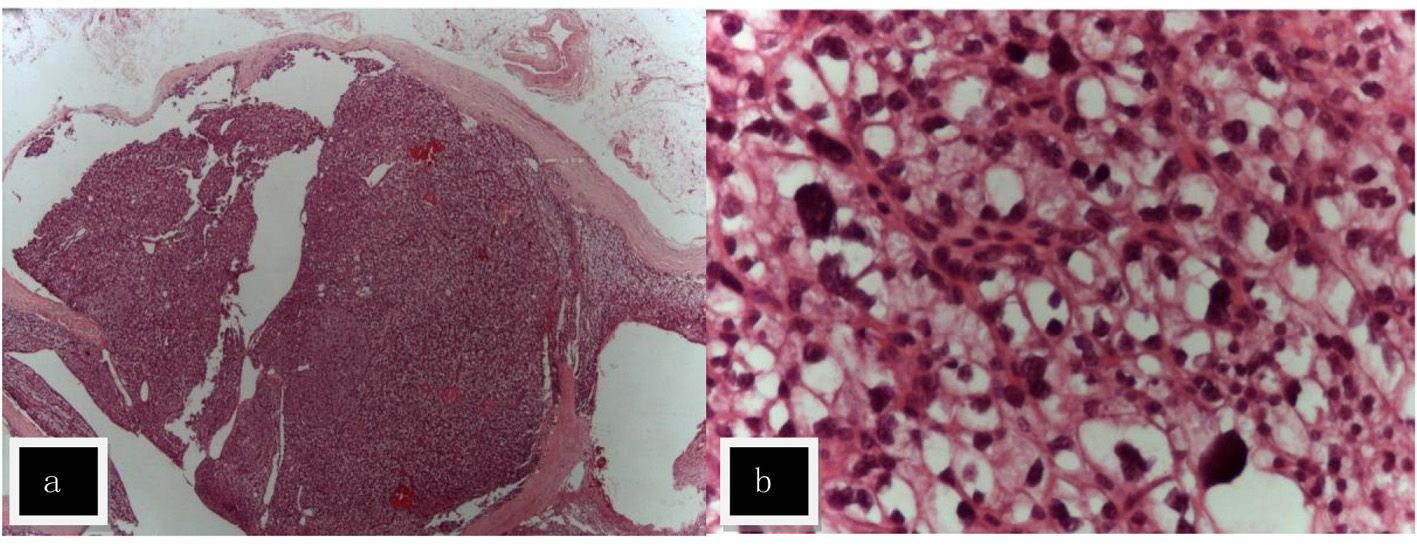 Click for large image | Figure 2. Hematoxylin and eosin staining (H&E) of atypical adenoma of the left parathyroid named (2) in the text. (a) The adenoma was surrounded by a fibrous capsule with septa (× 10). (b) The lesion was composed of nests of chief cells with clear cytoplasm and moderate cytological atypia (× 400). |
On October 2010, during routine surgical follow-up evaluation, the patient complained about a nodular lesion in the right thigh muscular mass that was present for several years, but had recently grown and was now slightly impairing the gait. A probable lipoma was assumed and that lesion was removed under local anesthesia. Instead an intramuscular non-smooth lesion was found 25 × 22 × 22 mm and pathological examination revealed a multinodular lesion with cystic and solid areas within muscular tissue. It was formed by confluent cells with abundant and clear cytoplasm, with numerous mitotic figures and marked cellular atypia with anisokaryosis and nuclear hyperchromatism (Fig. 3). The tumor extended to the surgical margin. There was strong immunoreactivity for vimentin, EMA, CD10 and calretinin, with rare cells positive for S-100, AE1/AE3, and focal immunoreactivity for PTH while no immunoreactivity was found for inhibin, actin, desmin, CK8/18, CK5/6, CK7, CK20, MNF-116, WT-1, melan-A, pCEA, FAPm, PSA, Hepar-1, chromogranin A and synaptophysin. In conclusion, data were suggestive of a metastatic carcinoma with clear cells with indeterminate origin, possibly kidney or adrenal. There was strong expression of the APC protein and focally positive parafibromin staining.
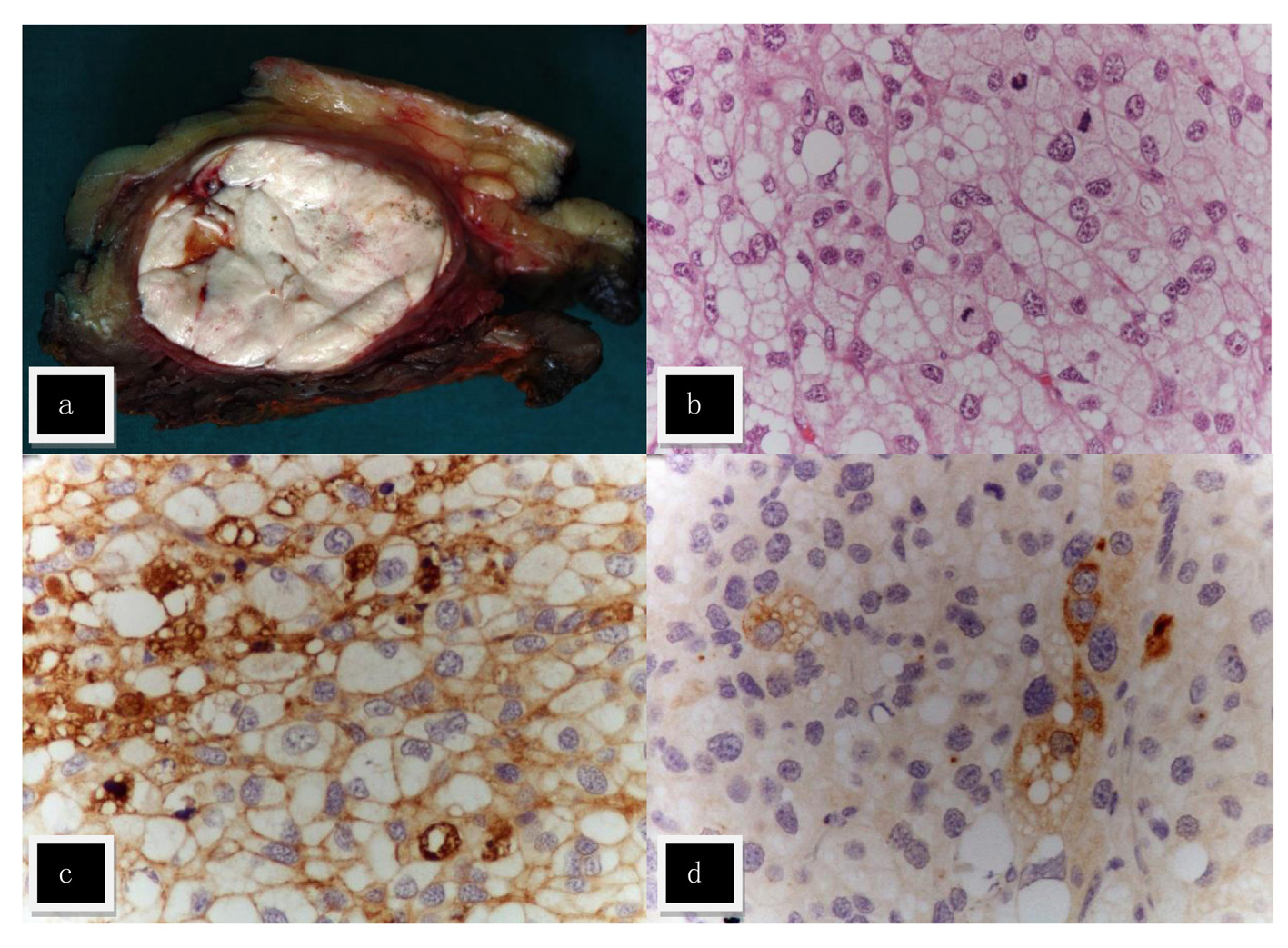 Click for large image | Figure 3. Metastasis in the right thigh. (a) Macroscopically the tumor was nodular, white and with a fleshy consistence. (b) The lesion was composed of cells with abundant clear and vacuolized cytoplasm, with marked cellular atypia; numerous mitotic figures were found (H&E, × 400). Focal immunostaining with cytokeratin cocktail AE1/AE3 (c) and PTH (d) was found. |
Endocrine re-evaluation on December 2010 evidenced no abnormalities of mineral metabolism or parathormone levels. Tumor markers like NSE, CA19.9, CEA, β-HCG and alpha-fetoprotein were negative. Genetic tests for MENIN, RET and HRPT2 mutations were negative. The following imaging methods were obtained: 1) CT scan - no cervical, thoracic and abdominal lesions suggestive of primary or metastatic disease were noted. A single simple renal cyst (< 1 cm) was present at the left kidney; 2) sestamibi scan - no cervical or mediastinal lesions were found; there was hyperfixation of the radionuclide at the outer side of the right thigh, more marked on earlier images with partial late resolution (Fig. 4a); 3) positron emission tomography (PET) with FDG-F18 - with normal distribution over the brain, salivary glands, heart, liver, kidney, bowel and bladder; enhanced fixation at the diaphragmatic suture (SUV 4.3) and at the right thigh, quadricipital area (SUV 1.8 at the first acquisition and SUV 2.6 at the second acquisition, 145 min after endovenous injection of FDG-F18).
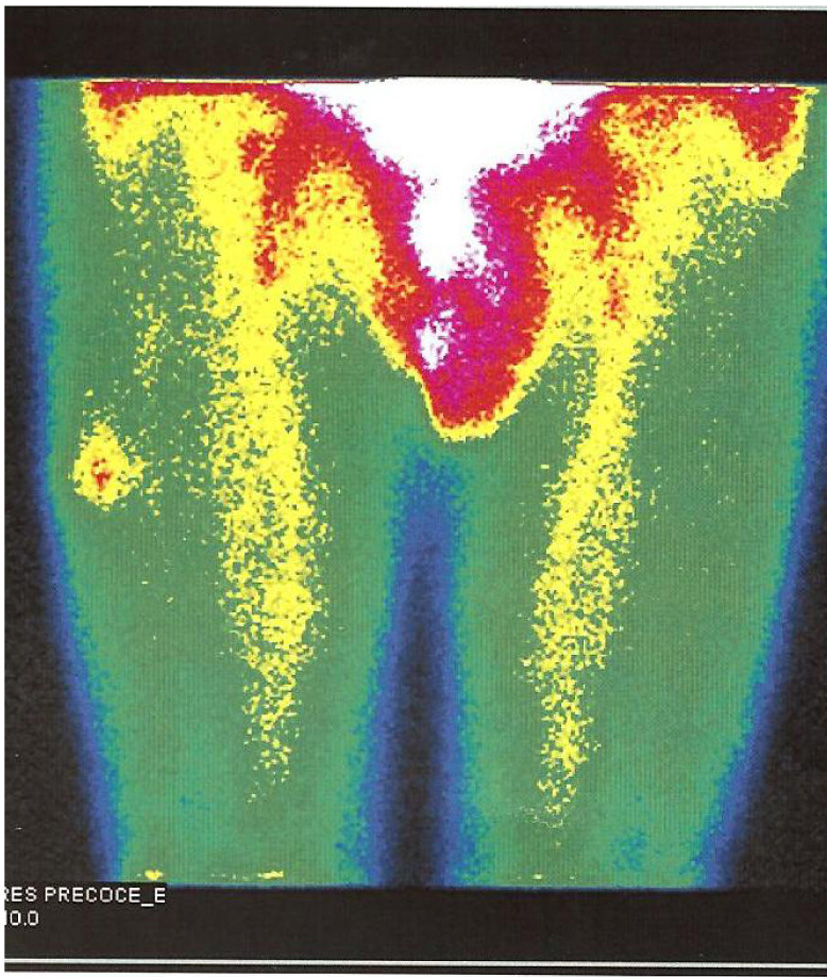 Click for large image | Figure 4. Sestamibi scintigraphy - hyperfixation of the radionuclide at the outer side of the right thigh. |
Surgical resection of the thigh mass was completed. The new sample was 17 × 7 cm and included a linear scar and three white-yellow lesions of 52, 20 and 17 mm, with well defined margins. Pathological evaluation revealed sparse cell with clear and microvacuolized cytoplasm with marked atypia, anisokaryosis and hyperchromatic nucleus, numerous mitosis; wide necrotic areas were identified; surgical margins were now free of neoplastic cells. Pathologic characteristics were similar to previously described.
After 2 years the patient remains clinically well, apparently free of disease, with normal mineral metabolism, and no evidence of renal or adrenal disease.
| Discussion | ▴Top |
This clinical case begins with a well differentiated gastric carcinoma of the intestinal type. Although it may be unrelated, it is tempting to speculate that long standing hypercalcemia, increased gastric acid secretion and subsequent chronic gastritis with intestinal metaplasia, may have eventually progressed to carcinoma. Although peptic ulcer disease is recognized as a possible complication of parathyroid malignant disease, we report a case in which the gastric malignancy was the first diagnosis. In a recent review, authors used the remarkable Swedish Family-Cancer Database comprising more than 12.2 million individuals, including more than 1 million cancer survivors, of which 87 with parathyroid carcinoma. Taking care to exclude cases included in MEN syndromes, and taking into account several potential confounding variables, the authors did not find any evidence for an increased risk for other cancer, namely gastric cancer, in patients with parathyroid carcinoma, and in relation to parathyroid adenoma only a small increased incidence of small intestinal cancer was found [5].
Signs and symptoms of hyperparathyroidism started 15 - 25 years before with HBP, calcium oxalate nephrolithiasis and nonspecific gastric complaints. Although individually either may be rather common, the combination is very suggestive of primary hyperparathyroidism [1, 2]. Benign primary hyperparathyroidism is also suggested by the mild hypercalcemia and mild increased serum PTH levels [1, 2]. However instead of the more common single adenoma (85%) that was also suggested by the sestamibi scan, two large adenomas were found with different pathologic characteristics: a right typical adenoma, recognized by sonography and sestamibi uptake and a left atypical adenoma with cystic degeneration, silent regarding isotope labeling and sonographic imaging but recognized by NMR. We could have thought about parathyroid carcinoma at this stage, namely because of patient gender, but as noted the clinical picture was against that hypothesis, imaging did not reveal any evidence for local or distant extension of the disease, pathological and genetic data are not unequivocal and furthermore the patient was apparently cured by surgery with complete normalization of mineral metabolism and PTH levels. Secondly the data suggest asynchronously evoluting adenomas, on the left probably earlier with cystic degeneration that may account for negative labeling and atypical aspects - thick capsule, and wide trabecula - and more recently on the right. A continuum hyperplasia - typical adenoma - atypical adenoma is well established in MEN and HPT-JT syndromes. Most emphatically Shattuck et al showed that parathyroid adenomas may occur asynchronously in the context of HPT-JT syndrome and Shane et al found some cases of carcinoma occurring in hyperplastic and adenomatous parathyroid glands and in one case of secondary hyperparathyroidism due to celiac disease [2, 6].
Most remarkably is the later presentation of a solitary metastatic lesion within the right thigh muscular mass. We are unaware of other similar reports [7, 8]. A gastric origin is a very remote possibility, given the well differentiated nature of that lesion, no evidence of local recurrence and the absence of other metastasis in more common locations as the liver. Furthermore the histological and immunohistochemical features of the thigh tumor were not suggestive of metastatic gastric adenocarcinoma.
We assume the metastasis to be of parathyroid origin, even if more common locations are the lung (40%), cervical lymph nodes (30%) and liver (10%): it uptakes sestamibi, although other primary mesenchymatous tumors may also label with the isotope; morphological and immunohistochemical features are that of a carcinoma, not sarcoma-like tumor; although the pathologists were emphatic regarding the possibility of a renal or adrenal tumor, no evidence of such a tumor was found by CT scan, PET imaging or scintigraphy and after 2 years there is still no evidence at all of that tumor. Besides the patient already has a known primary tumor, indeed two primary tumors; furthermore it would also be equally unlike for a renal or adrenal tumor to present as a single metastatic leg lesion. There remains the mystery of the unusual location of the metastasis; although we cannot prove it, the patient refers an old lesion at that location, and we assume that old lesion to be operative in the seeding of circulating neoplastic cells [9, 10]. Curiously enough, this metastatic lesion was functional in what regards sestamibi uptake but did not result on any PTH increase or hypercalcemia, despite its large dimensions.
The reported clinical evolution is strongly suggestive of a genetic defect; however, the most common reported abnormalities - MENIN, RET and HRPT2 - were specifically searched for and excluded in this case. Given the multitude of abnormalities described this finding is not totally unexpected. There was the distinct possibility of a HRPT2 mutation since the clinical syndrome more commonly includes parathyroid carcinoma and is associated with renal abnormalities - renal hamartomas, renal cell carcinoma, Wilms tumor and cystic kidney disease [2, 3, 6, 11, 12].
MENIN and RET mutations seem to be particularly associated with parathyroid hyperplasia and adenoma formation, although parathyroid carcinoma may occasionally occur. Like HRPT2 and APC mutations, these are constitutional abnormalities found in all somatic cells, and the recognized clinical syndromes are autosomal dominant.
More common are the acquired multiple genetic abnormalities found in the tumor cells, particularly PRAD1 gene (locus 11q13), an oncogene whose product is a cell cycle regulator, overexpressed in most (> 90%) of parathyroid carcinomas, or the tumor suppressor RB gene, whose product, is also a cell cycle regulator that is underexpressed in most of parathyroid carcinomas (100%). The tumor suppressor gene HRPT2, whose product is the parafibromin protein in also inactivated in most cases of the hereditary HPT-JT syndrome and in most cases (66%) of sporadic parathyroid carcinoma; parafibromin is present in several tissues besides the parathyroid, namely adrenal, kidney, heart and skeletal muscle, and although the mechanism of action remains unknown, it probably acts to inhibit cell proliferation and regulates transcriptional control [1-3, 6, 11, 13]. In fact a nuclear localization signal and three distinct nucleolar localization signals of parafibromin seem to be important for its function [14]. It has been postulated that parafibromin regulated expression of cyclin D1 [15]. Loss of immunoreactivity for parafibromin is highly sensitive and specific for the pathological diagnosis of parathyroid carcinoma [6, 10, 13, 14]. Inactivation of the APC tumor suppressor gene has also been implicated, since reduced expression of the APC protein is found in most parathyroid carcinomas [14]. The APC protein probably acts through the oncoprotein β-catenin [16]. These data have generally been interpreted according to the “two-hit” Knudson’s hypothesis of oncogenesis [1-3].
In short we describe a very unusual case of primary hyperparathyroidism first presenting with a gastric carcinoma, with two different parathyroid adenomas and with a late single metastatic lesion in the thigh. The nature of this last lesion remains undefined, since pathologically it resembles but not exactly either parathyroid lesions, contrariwise to these lesions it presents focal immunoreactivity for parafibromin, and although it uptakes the sestamibi isotope, there is no evidence for PTH secretion. However even if a comprehensive search was undertaken, no other primary tumor was found, namely renal, and again a single metastatic leg lesion of such a primary tumor would also be very unusual.
Disclosure
The authors have nothing to declare.
| References | ▴Top |
- Silverberg SJ, Bilezkian JP. Chapter 62, Primary hyperparathyroidism. In: Jameson JL, De Groot LJ, (senior editors), Endocrinology - Adult and Pediatric, 6th edition, Philadelphia: Saunders Elsevier; 2010. p: 1176-1197.
- Shane E. Clinical review 122: Parathyroid carcinoma. J Clin Endocrinol Metab. 2001;86(2):485-493.
doi pubmed - DeLellis RA. Parathyroid tumors and related disorders. Mod Pathol. 2011;24(Suppl 2):S78-93.
doi pubmed - Szmuilowicz ED, Utiger RD. A case of parathyroid carcinoma with hypercalcemia responsive to cinacalcet therapy. Nat Clin Pract Endocrinol Metab. 2006;2(5):291-296; quiz 297.
doi pubmed - Fallah M, Kharazmi E, Sundquist J, Hemminki K. Nonendocrine cancers associated with benign and malignant parathyroid tumors. J Clin Endocrinol Metab. 2011;96(7):E1108-1114.
doi pubmed - Shattuck TM, Valimaki S, Obara T, Gaz RD, Clark OH, Shoback D, Wierman ME, et al. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med. 2003;349(18):1722-1729.
doi pubmed - Gardner CJ, Wieshmann H, Gosney J, Carr HM, Macfarlane IA, Cuthbertson DJ. Localization of metastatic parathyroid carcinoma by 18F FDG PET scanning. J Clin Endocrinol Metab. 2010;95(11):4844-4845.
doi pubmed - Christofori G. New signals from the invasive front. Nature. 2006;441(7092):444-450.
doi pubmed - Chiang AC, Massague J. Molecular basis of metastasis. N Engl J Med. 2008;359(26):2814-2823.
doi pubmed - Howell VM, Haven CJ, Kahnoski K, Khoo SK, Petillo D, Chen J, Fleuren GJ, et al. HRPT2 mutations are associated with malignancy in sporadic parathyroid tumours. J Med Genet. 2003;40(9):657-663.
doi pubmed - Krebs LJ, Shattuck TM, Arnold A. HRPT2 mutational analysis of typical sporadic parathyroid adenomas. J Clin Endocrinol Metab. 2005;90(9):5015-5017.
doi pubmed - Howell VM, Gill A, Clarkson A, Nelson AE, Dunne R, Delbridge LW, Robinson BG, et al. Accuracy of combined protein gene product 9.5 and parafibromin markers for immunohistochemical diagnosis of parathyroid carcinoma. J Clin Endocrinol Metab. 2009;94(2):434-441.
doi pubmed - Juhlin CC, Haglund F, Obara T, Arnold A, Larsson C, Hoog A. Absence of nucleolar parafibromin immunoreactivity in subsets of parathyroid malignant tumours. Virchows Arch. 2011;459(1):47-53.
doi pubmed - Woodard GE, Lin L, Zhang JH, Agarwal SK, Marx SJ, Simonds WF. Parafibromin, product of the hyperparathyroidism-jaw tumor syndrome gene HRPT2, regulates cyclin D1/PRAD1 expression. Oncogene. 2005;24(7):1272-1276.
doi pubmed - Andreasson A, Sulaiman L, do Vale S, Martins JM, Ferreira F, Miltenberger-Miltenyi G, Batista L, et al. Molecular characterization of parathyroid tumors from two patients with hereditary colorectal cancer syndromes. Fam Cancer. 2012;11(3):355-362.
doi pubmed - Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275(5307):1787-1790.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.