

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website http://www.jofem.org |
Review
Volume 5, Number 5, October 2015, pages 273-283
Harnessing Structural Data of Insulin and Insulin Receptor for Therapeutic Designs
Suryani Lukmana, b, d, Habiba Al Safara, Sung Mun Leea, Kelvin Simc
aKhalifa University of Science, Technology and Research, Abu Dhabi Campus, PO Box 127788, Abu Dhabi, United Arab Emirates
bBioinformatics Institute, Agency for Science, Technology and Research, 30 Biopolis Street, #07-01 Matrix, Singapore 138671, Singapore
cInstitute for Infocomm Research, Agency for Science, Technology and Research, Singapore, 1 Fusionopolis Way, Singapore 138632, Singapore
dCorresponding Author: Suryani Lukman, Khalifa University of Science, Technology and Research, Abu Dhabi Campus, PO Box 127788, Abu Dhabi, United Arab Emirates
Manuscript accepted for publication August 14, 2015
Short title: Insulin and Its Receptor Structure Review
doi: http://dx.doi.org/10.14740/jem302w
- Abstract
- Introduction
- Structural Features of Insulin
- IR: A Membrane Glycoprotein
- Conformational Dynamics and Allostery Facilitate the Insulin-IR Interaction
- The Specific TM Domain of IR
- Literature Mining of Insulin and Its Receptor
- Subtle Differences of IR Isoforms in Molecular Functions
- Structural Data for Proposing Potential Therapeutic Strategies
- Nascent Nanotechnology-Based Approaches for Controlled Delivery of Insulin
- Conclusion
- References
| Abstract | ▴Top |
To lower blood glucose concentration, insulin binds to insulin receptor (IR) that possesses two distinct insulin binding sites to trigger downstream signaling events leading to an increased uptake of glucose into muscle and fat cells. Comprehensive understandings of structural and dynamic mechanisms of insulin and its receptor are essential to design therapeutic agents for treating and delaying the onset of diabetes that affects over 347 million people worldwide. No full-length IR structure is available hitherto. Harnessing the currently available and state-of-the-art sequence and structural data, we have reviewed the insulin, IR, its extracellular domains and transmembrane domain, to derive structure-based clues to regulate aberrant insulin and its receptor. To propose testable hypotheses and future experiments, we have performed literature review, text mining, multiple structural clustering and normal mode analysis on insulin and its receptor. It appears that insulin-receptor interaction involves allostery and conformational changes (including rotation and tilting) to overcome steric clashes. To target a particular aberrant isoform of IRs, we need to identify the subtle yet distinct differences between IR isoforms. To improve the life quality of diabetics, better structure-based designs of insulin mimetics, formulation and nanotechnology-based delivery are required; efforts to bring them to patients necessitate thorough structural understandings of insulin and its receptor.
Keywords: Insulin; Insulin receptor; Structural analysis; Allostery; Structure-based drug design
| Introduction | ▴Top |
Insulin, first isolated in 1921, was soon deployed as a trial therapeutic to save the lives of diabetic patients in 1922. Since then, the lives of millions of diabetes patients have been saved and prolonged. The sequence of insulin was first determined by Fred Sanger in 1955 and its structure was first resolved by Dorothy Hodgkin in 1969.
The overall action of insulin is energy storage by the body, as opposed to energy consumption, which is the overall action of glucagon. The synthesis and secretion of insulin are regulated by the blood glucose concentration. To store energy, insulin lowers blood glucose by promoting 1) the uptake of glucose into cells, particularly muscle and fat cells that express GLUT4 glucose transporter and 2) the storage of glucose as glycogen or fat.
In this review, we discuss how structural features of insulin highlight the importance of conserved residues, the structural domains of insulin receptor (IR), how IR relays insulin signals, the conformational dynamics and allostery that facilitate the insulin-IR interaction, the differences between IR isoforms, the molecular functions of insulin and related hormones, and testable hypotheses useful for therapeutic designs targeting insulin and IR.
| Structural Features of Insulin | ▴Top |
Pancreatic cells synthesize insulin in the form of preproinsulin. The removal of an N-terminal signaling peptide of preproinsulin (110 residues), on translocation into endoplasmic reticulum, results in proinsulin (86 residues). Proinsulin is cleaved into chain B (30 residues of FVNQHLCGSHLVEALYLVCGERGFFYTPKT) and chain A (21 residues of GIVEQCCTSICSLYQLENYCN) by β-cells peptidases (Fig. 1), during the trafficking through the trans-Golgi network. Before the cleavage, the conserved chains B and A are linked by a non-conserved connecting domain, also known as the C-peptide. After cleavage, the resulting heterodimer of chains A and B consists of 51 amino acids (molecular weight is 5,808 Da) and is still linked by two disulfide bonds: Cys7 of chain A and Cys7 of chain B, Cys20 of chain A and Cys19 of chain B. Within chain A, a disulfide bridge links the Cys6 and Cys11 (Fig. 1). These Cys residues that form the three disulfide bonds are conserved across species and members of insulin superfamily; these Cys residues play critical roles in conferring a similar overall tertiary structure. Insulin mutations that introduce or remove a Cys, perturb the native disulfide bonds. These native bonds are ensured through the proper folding of proinsulin, and its misfolding has been linked to neonatal diabetes mellitus-associated mutations [1]. The processing of proinsulin, in terms of cleavage site selection (at Arg55-Arg56 and Lys88-Arg89), relies on the flexibility of both ends of the C-peptide. The presence of the C-peptide in proinsulin renders it to be more resistant to misfolding, aggregation and fibril formation than insulin [2]. The pathogenic fibril formation is cytotoxic, present in over 90% of type 2 diabetes, and causing death of insulin-secreting pancreatic β cells [3].
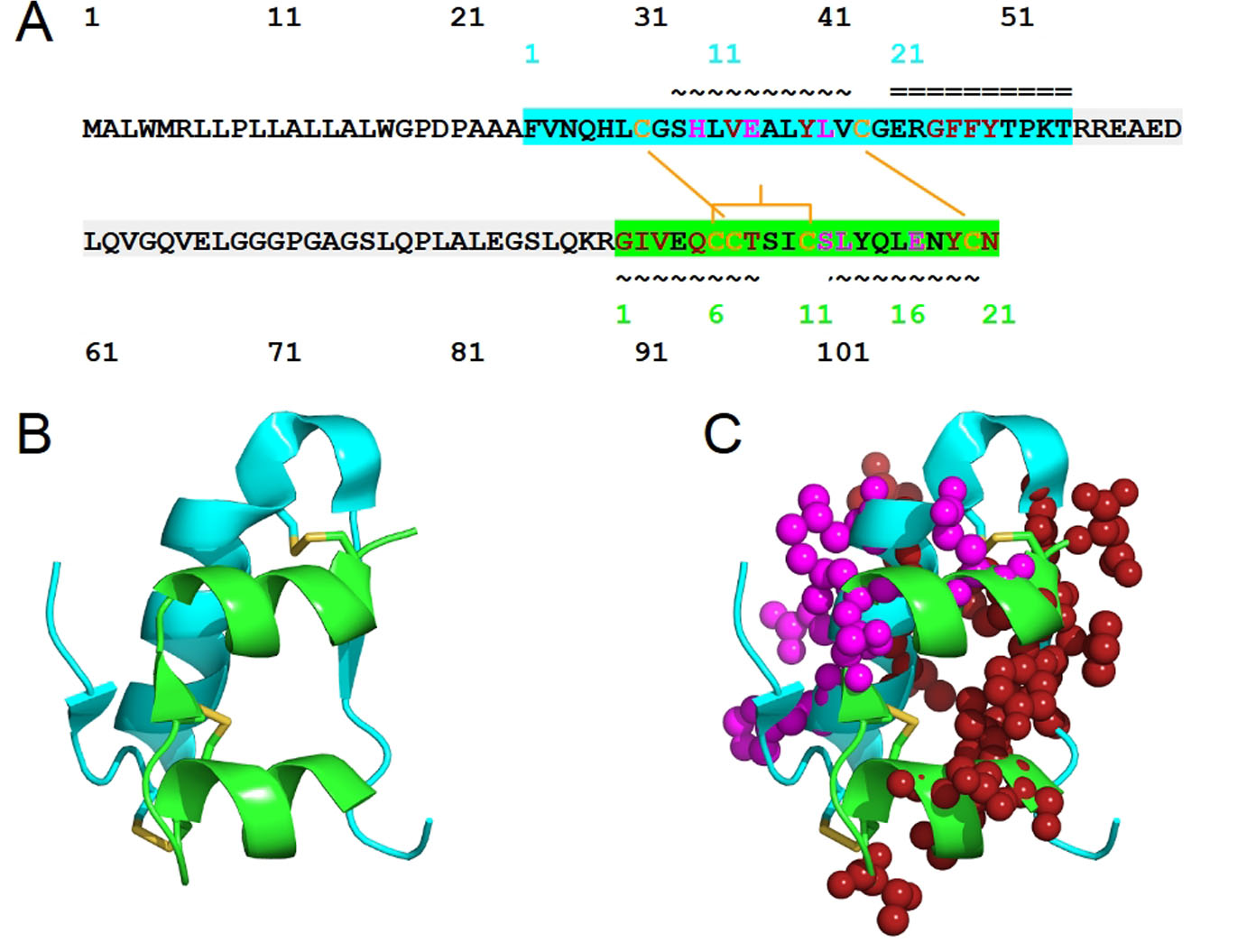 Click for large image | Figure 1. The structure of human insulin. (A) The sequence of human insulin in the form of preproinsulin (UniProt ID P01308 numbering in black) is highlighted according to the respective chains of insulin: chain B (cyan) and chain A (green). C-peptide is highlighted in light gray. Cysteine residues involved in inter-chain disulfide bridges (Cys7 of chain A and Cys7 of chain B, Cys20 of chain A and Cys19 of chain B) and those that contribute to intra-chain A disulfide bridge, Cys6 and Cys 11, are shown in yellow-orange. The secondary structure of human insulin consists of three alpha-helices formed by chain A residues Gly1-Thr8, Ser12-Asn18 and chain B residues Ser9-Cys19. The residue numbers of insulin chains B and A are shown in cyan and green, respectively. The symbols ~ and = indicate α-helices and β-strands, respectively. Insulin residues that bind to IR sites 1 and 2 are shown in red and magenta, respectively. (B) The structure of human insulin with the same coloring scheme as (A) and the insulin residues that bind to IR sites 1 and 2 are shown in spheres (C). |
In terms of secondary structure, insulin consists of three alpha-helices formed by chain A residues Gly1-Thr8, Ser12-Asn18 and chain B residues Ser9-Cys19 [4]. Inactive insulin is zinc-ion-bound hexameric [5] - the form insulin is produced and stored. Depending on the zinc-mediated stabilization, insulin hexamer can adopt T6, T3R3 or R6 structure; their T-R transition is regulated through allostery, an essential consideration for the design of therapeutic insulin. Active and circulated insulin is monomeric. Dimeric insulin [6] is also present when its concentration reaches micromolar level; insulin dimerization requires about -7.2 kcal/mol and is driven by vibrational entropy [7].
| IR: A Membrane Glycoprotein | ▴Top |
Encoded by 22 exons [8], IR is a disulfide-linked membrane glycoprotein comprising two alpha-subunits and two beta-subunits, all together forming two half-receptors. The alpha-subunits form the hormone binding domains, also known as ectodomains. In this work, we refer to the residues of full-length IR according to the numbering of UniProt ID P06213.
There is no full-length experimentally determined IR structure hitherto, due to the highly flexible nature of the IR domains. The structure of an IR half-receptor comprises leucine-rich L1 and L2 domains, spanned by cysteine-rich (CR) domain, type III fibronectin domains (Fn1, Fn2, and Fn3), an insert domain (IR) within the Fn2 domain [9], a transmembrane (TM) domain, a tyrosine kinase domain and a C-terminal tail (Fig. 2). To form a half-receptor, an alpha-subunit and a beta-subunit are linked by a disulfide bridge, which is formed between residues Cys674 in the alpha-subunit and Cys899 in the beta-subunit [10]. The extracellular alpha-subunits are linked to each other by a disulfide bridge between residues Cys551 [11]. The L1-CR-L2 domains of a half-receptor pack against the Fn1-Fn2-Fn3 domains of another half-receptor [4], forming a folded-over conformation akin to St. Louis Gateway Arch (Fig. 2).
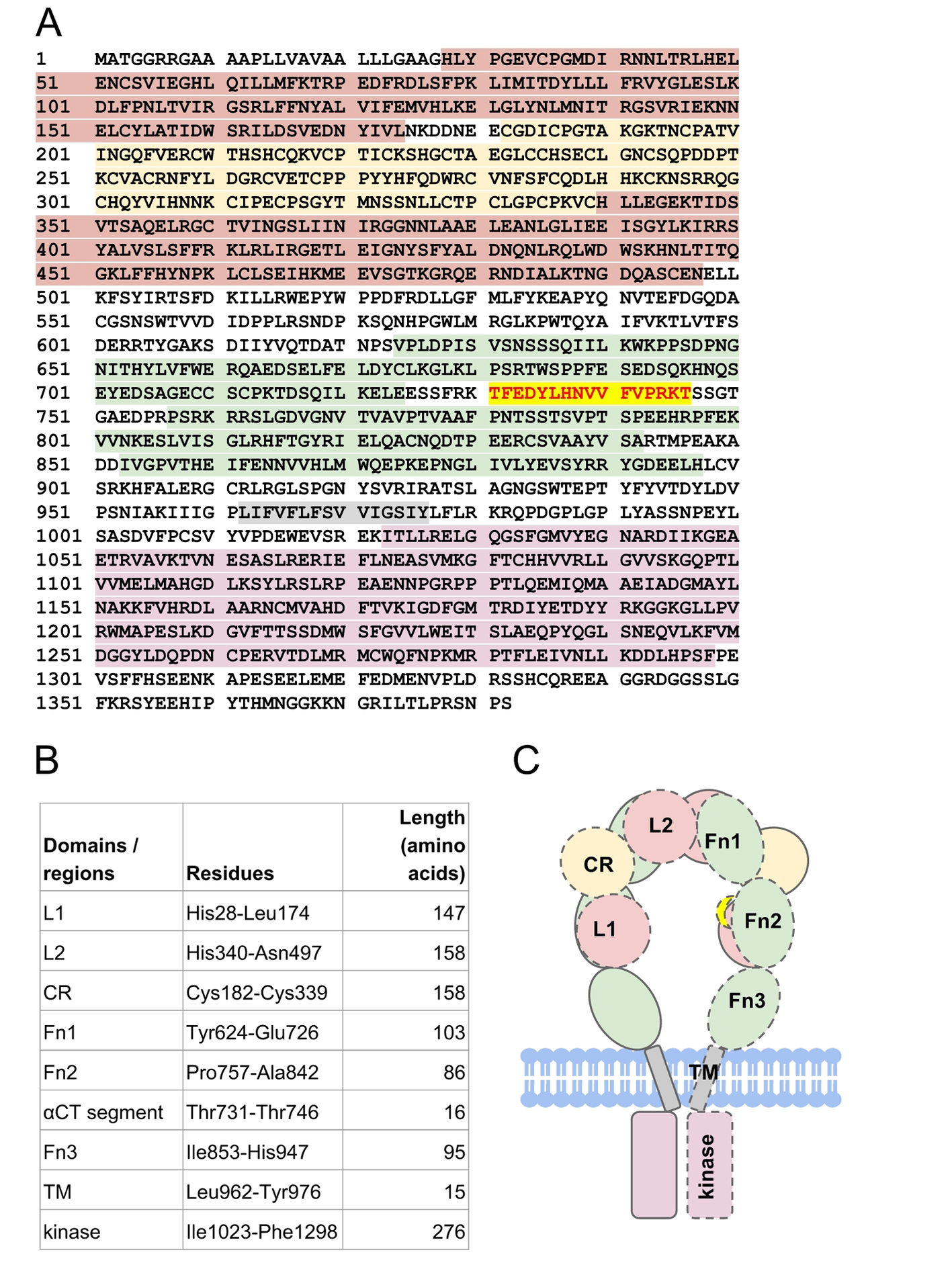 Click for large image | Figure 2. The structure of human insulin receptor. (A) The sequence of human insulin receptor (UniProt ID P06213) is highlighted according to its respective domains (B): leucine-rich domains (red maroon), cysteine-rich domain (yellow), type III fibronectin domains (green), transmembrane (gray) domain, and tyrosine kinase domain (pink). Residues Thr731-Thr746 (red scarlet) forms the αCT segment that is important for insulin binding. (C) Schematic representation of insulin receptor domains in the absence of insulin. The domains of a half-receptor and the other half-receptor of insulin receptor are distinguished by solid and dashed lines. |
IR relays signal from insulin to intracellular signaling through phosphorylations of proteins including insulin receptor substrate (IRS) family members (IRS-1, IRS-2, etc.), Shc and Cbl. The phosphorylated forms of these proteins act as docking sites for proteins with SH2 domains that specifically detect the phosphotyrosine residue on the short linear motif YXXX [12]. These proteins containing SH2-domain(s) - for example PI3K, GRB2 and SHP2 - are critical for diverse signaling outcomes.
| Conformational Dynamics and Allostery Facilitate the Insulin-IR Interaction | ▴Top |
In the absence of insulin, both IR half-receptors are held in a constrained position. These constraints are released upon the binding of insulin. The binding of insulin to the alpha-subunits triggers a conformational change on the beta-subunits, auto-trans-phosphorylation of the tyrosine kinase domains of IR, and a series of signaling events involving multiple phosphorylation and dephosphorylation [13].
Although there are two insulin epitopes (binding sites) of distinct interfaces [14] on IR, the molecular details of insulin binding to IR have remained largely elusive. The pioneer and breakthrough X-ray crystallographic structures of the complex between insulin and truncated IR, without the majority of Fn domains, were resolved in 2013 [4]. We note that both interacting partners, in the complex structure of PDB entry 3W14, are from distinct species: bovine insulin and human truncated IR. The complex structures show that the insulin normally binds to only a side of the symmetrical IR and predominantly binds to the C-terminal of α-chain, abbreviated as αCT segment that corresponds to residues Thr704-Thr719 of an IR half-receptor; this binding displaces the C-terminal β-strand (of insulin B-chain) away from the core of insulin [4].
It is not only the insulin that undergoes conformational changes, the αCT segment also repositions the L1 domain of IR; this is an interesting example of synergistic structural dynamics and allostery in ligand-receptor recognition and interaction. Both the αCT segment and a part of the L1 domain (non-polar residues Leu63, Leu64, Leu89, Phe91, Phe105, Phe106, Val121, and Phe123) that interacts with insulin are referred to as the binding site 1. Based on these observations, it is likely that insulin and insulin-mimetic peptides act similarly to the αCT segment [15]. It is also worth mentioning that the binding site 1 of insulin is formed by the αCT segment and L1 domain of different α-chain from another half-receptor. There are no known homologs for the αCT segment in other mammalian tyrosine kinase [16].
Binding site 2 of insulin consists of the loops at the junction of Fn1 and Fn2 domains in the other half-receptor [14]. How insulin binds to its binding site 2 remains largely unknown, and it may require conformational changes that displace both Fn1 and Fn2 domains away from the L1 and CR domains [4]. Based on a computational model generated using molecular dynamics flexible fitting [17], chains A and B residues of insulin are predominantly interacting with residues of Fn1 and Fn2 domains, respectively [18]. For example, insulin chain A residues Ser12, Leu13, Tyr14, Gln15, and Glu17 were predicted to interact with IR residues Arg581, Gly582, Leu583, Lys584, and Tyr589 [18]. Future scanning mutagenesis studies are essential to verify these interacting residues and to identify novel ones.
Differences exist for both insulin binding sites on IR. The binding affinity of insulin for binding site 2 (Kd about 400 nM) is lower than that for binding site 1 (Kd about 6.4 nM); the dissociation constants were obtained based on the kinetics fitting of harmonic oscillator model of IR, using genetic algorithm, to the experimental data [19]. To bind to the binding site 1, insulin uses its hormone-dimerizing residues (B-chain helix); whereas to bind to binding site 2, insulin uses its hormone-hexamerizing residues (A-chain residues) [20]. Such a distinct mechanism can potentially be extended to the insulin-mimetic peptides with therapeutic potentials.
Although the insulin binding sites 1 and 2 on IR are distinct, it appears possible that two insulin molecules bind simultaneously to a single IR [18]. The stoichiometry of insulin-IR binding is dependent on concentration. At picomolar concentrations, insulin binds to IR in a 1:1 ratio [19]. At higher nanomolar concentrations, two insulins bind to an IR and display a negative cooperativity [21], in which the binding of an insulin to a site on IR decreases the affinity of another insulin for the other site. The negative cooperativity is possibly achieved through the coupling between 1) the closing of Fn1-Fn2 hinge and L1-L2 hinge and 2) the opening of L2-Fn1 hinge [18], akin to a folded-over see-saw mechanism [9].
Not much is known regarding the sequential mechanism of insulin binding to both binding sites 1 and 2 of IR. Kinetic studies using soluble truncated IR suggest that insulin binds to site 2 very rapidly, followed by a slower rate-limiting conformational changes that enable the binding of insulin to site 1 [22]. While this study suggests a biphasic phase of insulin binding to soluble truncated IR, whether the same mechanism applies to full-length, membrane-bound IR remains to be verified.
It is important to note that the insulin-IR complex structures (PDB entry 3W14) do not represent the complete picture. Its insulin chain B contains only residues H5-E21 [4] and is shorter than the full-length insulin chain B of 30 residues. Chain B residues, such as Phe24, Phe25 and Tyr26 that dock on the IR, are missing. To resolve the coordinates of insulin chain B residues Arg22-Thr30, native Fn3 domain may be required in the construct [4]. When we replaced the truncated insulin (PDB 3W14) with the full-length insulin (PDB 4EY1), we observed a steric clash between the aromatic residues of insulin and the αCT segment of IR (Fig. 3), suggesting that conformational change, which is likely due to induced fit effects, and structural dynamics have to take place to avoid the steric clash.
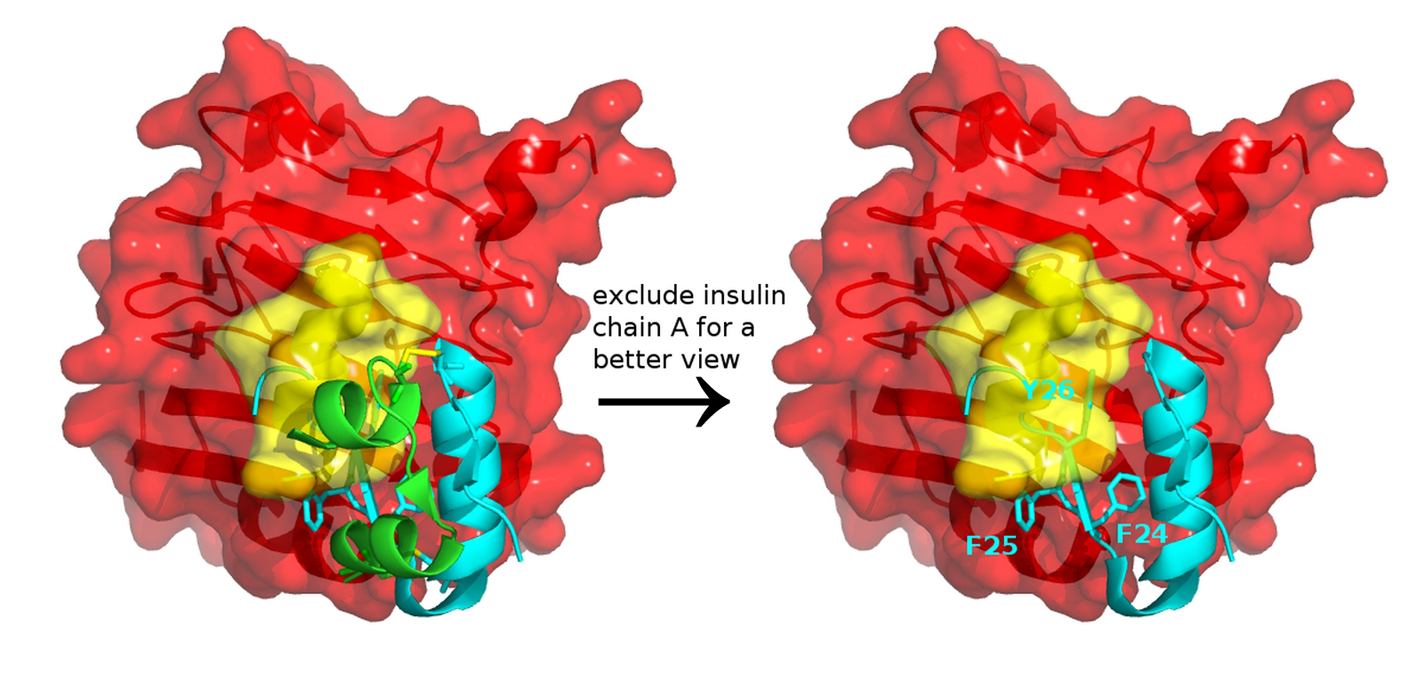 Click for large image | Figure 3. Insulin and insulin receptor interaction. Docking a full-length insulin (PDB entry 4EY1) to the IR structure (PDB entry 3W14) containing αCT (yellow) and L1 domain (red) highlights that conformational change and structural dynamics have to take place to avoid a steric clash between the aromatic residues of insulin and the αCT segment of IR. |
The insulin-truncated IR complex structures [4] also suggest that the insulin occupies the same overlapping region as the Fn1 and Fn2 domains do in the insulin-free (apo) full-length IR structure. The suggested steric clashes mean that the Fn1 and Fn2 domains need to undergo conformational changes, such as tilting, to allow the binding of insulin to IR. Conformational changes as a mechanism for ligand binding is relatively less studied among the family members of receptor tyrosine kinases [4], but conformational changes and allostery have been shown to be critical in facilitating both conserved and specific functions of diverse proteins such as Ras [23].
The globular insulin undergoes conformational changes for binding to its IR, yet the mechanisms have long been unclear until recently. In a study investigating the binding of the C-terminal insulin chain B (residues Phe24-Thr30) to a truncated IR comprising the primary hormone-binding site (L1 domain and αCT), the chain B peptide undergoes a concerted hinge-like rotation of about 10° at its β-turn, formed by residues Gly20-Gly23 [24]. In a coupling manner, the hinge-like rotation leads to an about 50 - 60° rotation (about the highly conserved Phe24 of chain B) of the β-strand formed by residues Phe25-Pro28 of chain B. The β-strand hence moves away from the insulin α-helical core 1) to lie anti-parallel to the β2 sheet of L1 domain of IR and 2) to enable the conserved aromatic motif of Phe24, Phe25, Tyr26 of chain B to dock between L1 and αCT. Consequently, the hinge-like rotation exposes the otherwise-buried (and conserved) hydrophobic residues of chain A (Ile2 and Val3) and chain B (residues Val 12, Phe24 and Phe25) for interaction with IR. Based on these proposed mechanisms, the substitution of Phe25Leu in “insulin Chicago” that results in a rare form of diabetes [25] can be rationalized as a steric clash with the αCT that reduces the “insulin Chicago” affinity to IR. Moreover, the aromatic ring of Phe24 is essential to anchor insulin to IR, through long-range packing interactions in the hydrophobic core [26]. The importance of residue Phe24 is highlighted by the Phe24Gly mutant that shows higher flexibility in the C-terminal of chain B, than the wild-type insulin do, as indicated through explicit-solvent molecular dynamics (MD) simulations of monomeric and dimeric insulins [27].
| The Specific TM Domain of IR | ▴Top |
Upon the binding of insulin to the alpha-subunit of IR, the TM domain of IR appears to be critical in relaying the insulin-induced conformational changes to the beta-subunits. An NMR structure (PDB code 2MFR) of IR TM domain (residues Leu962-Tyr976) shows that TM domain adopts an alpha-helix with a kink contributed by residue Pro961 [28]. Peptides mimicking TM domain residues can specifically activate wild-type IR and IR Ser323Leu mutant associated with insulin resistance, but not related growth factor receptors [29]. This specificity is achieved through the relatively low sequence identity of the TM domain between IR and other related growth factor receptors. The facts that the IR Ser323Leu mutation is situated on the CR domain, and the mimetic of TM domain can activate this IR mutant, imply a possible allostery between the CR and TM domains.
In the absence of insulin binding, the TM domains of IR form an obligate homodimer. The conformational switch induced by insulin binding [14], coupled with the obligate nature of the IR oligomerization [13], play critical roles in the trans-phosphorylation of the two tyrosine kinase domains of IR. This mechanism is different from the dimerization of many other receptor tyrosine kinases that can only occur upon ligand binding, in which the dimerization facilitates the trans-phosphorylation of one cytoplasmic by the other, and vice versa [30].
An IR-TM peptide [29] is proposed to activate IR through its interactions with each of the two TM domains of an IR, hence dissociating the TM domain homodimer, which is similar to the effect of insulin binding to the alpha-subunit of IR. The dissociation of the TM domains triggers conformational changes in allosteric manners, possibly including rotations that juxtapose the cytoplasmic kinase domains for their trans-phosphorylations.
| Literature Mining of Insulin and Its Receptor | ▴Top |
Through text mining of 2,901 literature abstracts of insulin hormones (Fig. 4), we discovered that there have been numerous studies on insulin in relation to other hormones, including glucagon, leptin, and cortisol. Glucagon increases blood glucose when the blood glucose levels decrease beyond normal. Leptin is the satiety hormone that inhibits the feeling of hunger once the amount of fat stored in adipose cells is sufficient. The effect of leptin is opposite to that of ghrelin, the hunger hormone. Both leptin and ghrelin act on the same brain receptors to regulate the energy metabolism in the body. In response to stress, cortisol, a steroid hormone, increases blood glucose and inhibits immune system. In brief, glucagon, cortisol and ghrelin can be classified in a group in terms of their physiological effects, whereas insulin and leptin can be classified in another group.
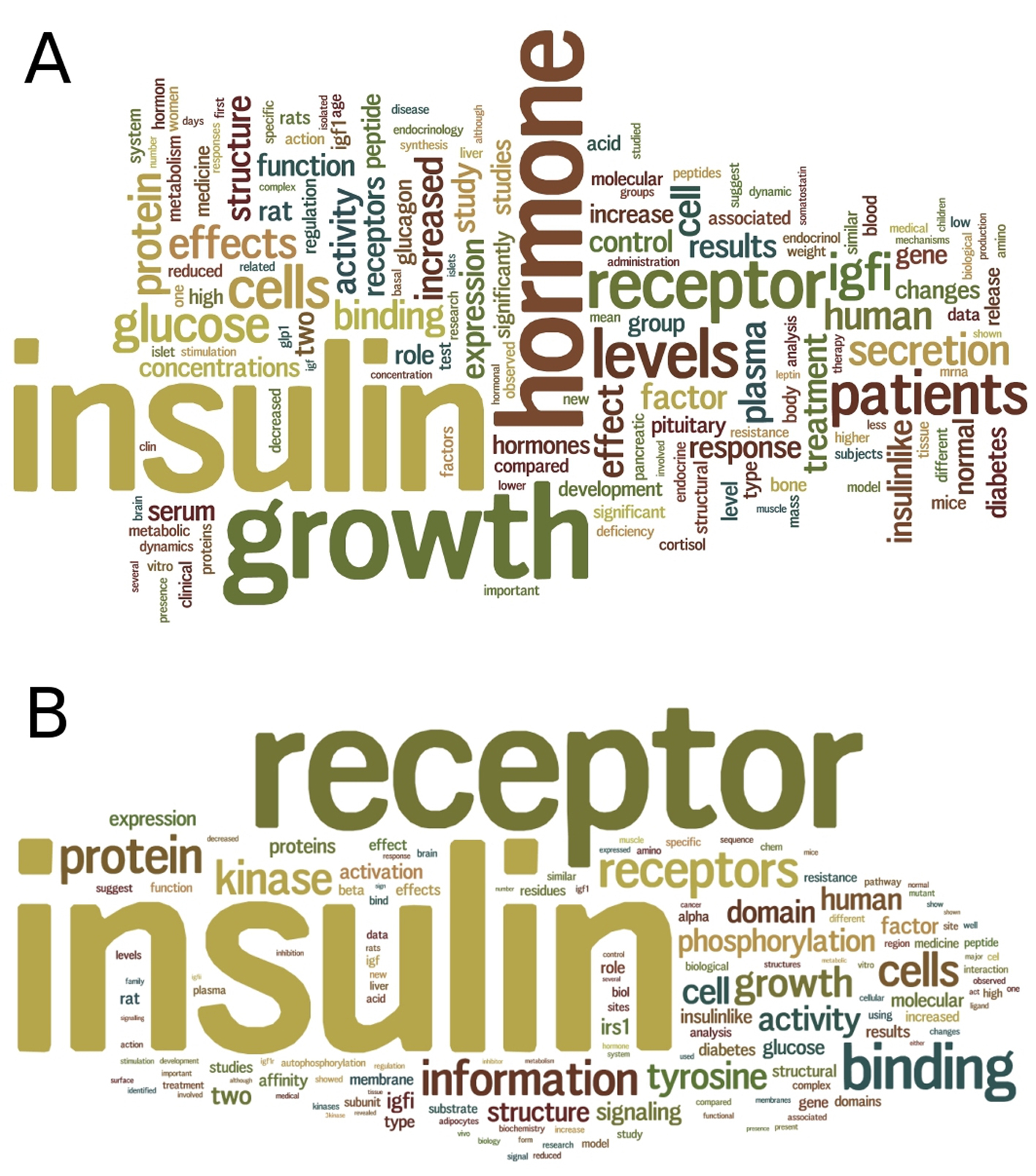 Click for large image | Figure 4. Text mining of insulin and insulin receptor literature. (A) A summary wordcloud generated from text mining of 2,901 abstracts of articles in PubMed for insulin hormone, provides a clue on the relationships of insulin with other hormones including glucagon, leptin and cortisol. (B) A summary wordcloud generated from text mining of 2,201 abstracts of articles in PubMed for insulin receptor, provides a clue on the binding of not only insulin but also insulin growth factor (IGF) to the insulin receptor. |
Through text mining of 2,201 literature abstracts of IR (Fig. 4), we discovered that there have been numerous studies on insulin-like growth factors (IGFs), such as IGFI and IGFII that share 36% and 41% of sequence identity to insulin, respectively (Fig. 5). We performed multiple sequence alignment of their sequences using Clustal Omega [31] and observed two relatively conserved regions spanning residues Leu30-Tyr50 and residues Gly90-Cys109 of the 110aa-long insulin sequence (Fig. 5); the two regions correspond to the cleaved chains B and A, respectively. In addition, all three hormones have the conserved Cys residues that form three disulfide bonds, contributing to their tertiary structural similarity. These similarities in sequence and structure of the hormones may partly explain the promiscuity of IR as the receptor of not only insulin but also IGFs.
 Click for large image | Figure 5. Sequence similarity among insulin and IGFs. (A) Pairwise sequence identity (%). (B) Multiple sequence alignment performed using Clustal Omega shows two relatively conserved regions (boxed) spanning residues L30-Y50 and residues G90-C109 of insulin hormone, which correspond to the cleaved chains B and A, respectively. |
Besides IGFs, Ahsg (fetuin-A) also binds to IR, particularly to its Fn3 domain [32]. Ahsg is a 55 - 59 kDa phosphorylated glycoprotein synthesized predominantly in liver cells and secreted to blood circulation. In contrast to insulin that activates the IR tyrosine kinase, Ahsg inhibits the IR tyrosine kinase and is present abundantly in the blood of people with insulin resistance and diabetes [33].
Physiologically, insulin and IGFs function to activate their respective receptor tyrosine kinases, leading to nearly identical signaling pathways [8], but remarkably distinct outcomes: insulin is responsible for metabolism, and IGFs induce mitosis [34]. IGFI promotes cell proliferation to attain maximal growth and inhibits apoptosis (cell death); IGFII promotes growth during early (placental and fetal) development. In disease states, IGFI and IGFII are associated with cancers. Besides binding to IGFI and IGFII receptors, IGFs also bind to IR [9]. Since insulin and IGFs utilize almost identical machineries and signaling pathways, the distinct mechanisms of how they achieve signaling specificity are intriguing. Their signaling specificity may have arisen due to differential attributes and wiring of overlapping protein network. To investigate such a network, a system biology approach can be pursued [35].
| Subtle Differences of IR Isoforms in Molecular Functions | ▴Top |
Alternative splicing of IR results in two isoforms: short IR-A (1,370 amino acids) and long IR-B (1,382 amino acids). The alternative splicing of exon 11 [8] results in IR-A isoform that misses residues Lys745-Arg756 of the canonical IR-B isoform. We observe a subtle difference in sequence, between the αCT segment of IR-A (residues T731FEDYLHNVVFVPRPS746) and IR-B (residues T731FEDYLHNVVFVPRKT746), specifically at the last two residues; this difference may contribute to subtle difference of insulin binding to IR isoforms, in relation to the crucial role of the αCT segment in forming the insulin binding site 1.
While both IR-A and IR-B isoforms do not vary greatly in their affinity for insulin, the IR-A isoform has higher affinity for IGFI and IGFII [36]. IR-A is also implicated in malignant transformation, in which IR-A is over-expressed in cancer cells that overexpress IGFII [37]. The complexity of these cross-talks between insulin-like growth hormones and IR, their mutations and implications on cancer and diabetes [38], have incessantly posed new challenges in the preventive and therapeutic efforts against these diseases.
Soluble ectodomains of IR-A are shown to be more abundant in the blood plasma of diabetic patients [39] and may be used as a rapid glycemic marker. How effective the soluble IR-A ectodomains as a marker (as compared to A1C or glycoalbumin) remains to be verified. The cleavage mechanisms of ectodomains from the membrane-bound IR-A are also largely unclear, and the (unknown) protease responsible for the cleavage may be activated by hyperglycemia in diabetes. The latest clue is that O-linked N-acetylglucosamine modification activates IR-A ectodomain cleavage by an extracellular calcium-dependent protease [40]. These findings lead to the following basic scientific questions. What is the protease? Where (which residues) does the protease cleave on IR-A? What are the mechanisms of the proteolysis? Does the increase of soluble ectodomains of IR-A in blood plasma deprive insulin from binding to IR for insulin signaling, implying the aberrant nature of soluble IR-A ectodomains?
To identify the protease responsible for cleaving IR-A ectodomain, fluorimetric assays and immunoblotting can be performed. An indirect evidence supporting our hypothesis is that the higher the plasma level of soluble IR ectodomains is, the insulin resistance becomes more severe in diabetic patients [40]. An applied science question worth pursuing is how to selectively sequester soluble IR-A ectodomains using small molecules or peptide mimetics (e.g. stapled peptides and cyclic peptides) that will bind only to the soluble IR-A but not bind to the membrane-bound IR, so that there will be sufficient amount of insulin available to bind to membrane-bound IR to propagate insulin signaling.
Metformin, a biguanide small molecule with an anti-hyperglycemic effect, was found be useful to prevent the IR-A ectodomain cleavage [40], yet the atomistic mechanisms remain unresolved. In terms of molecular mechanisms, metformin inhibits mitochondrial respiratory chain complex I [41], but its exact mechanism(s) remain to be discovered. While metformin is commonly prescribed as oral diabetes medicine to over 100 million patients worldwide [42], it can lead to the life-threatening lactic acidosis in people with liver/heart diseases.
| Structural Data for Proposing Potential Therapeutic Strategies | ▴Top |
Mutations of IR are associated with multiple pathological conditions, including Rabson-Mendenhall syndrome (RMS), Donohue syndrome/leprechaunism, and insulin-resistant diabetes mellitus [43]. The mechanisms of IR mutations include impaired insulin binding to the IR and impaired tyrosine kinase activity. Thorough examination of the effects of these first mutations on the structures and structural dynamics of IR can suggest suppressor (second) mutations that alleviate or reverse the effects of the first mutations, in which the suppressor mutations act locally or allosterically through hydrogen-bond networks and/or electrostatic forces. For example, pairs of distant residues that share correlated motions can potentially be pair candidates of first and suppressor mutants [44]. Identification of suppressor mutants in silico is more cost-, labor- and time-efficient than high throughput screening of libraries of compounds.
Multiple structures determined by diverse laboratories under different conditions can provide a glimpse of the structural dynamics of the protein of interest. Using structural clustering methods such as principal component analysis [45] and subspace clustering [46], we have determined the representative structure of multiple parts of IR structures (Fig. 6), to provide clues for subsequent studies, including MD simulations and molecular engineering. For residues Pro31-Lys337 of IR that form the L1 and CR domains, the clustroid (representative) structure of 10 PDB structures is 3LOH_E that comprises residues Pro31-Ser682. For residues Asp1014-Lys1310 of IR, encompassing the tyrosine kinase domain, the clustroid structure of 20 PDB structures is 3BU3_A. Knowledge on the intrinsic structural dynamics of biomolecules can be used to design therapeutic strategies to target aberrant conformational switches [47]. For example, probing the structural dynamics of HIV integrase has facilitated the discovery of a novel binding site that opens only transiently, yet presents a higher affinity once ligands bind to it [48]. The discovery has led to the first FDA-approved drug that inhibits HIV integrase for the treatment of AIDS [49].
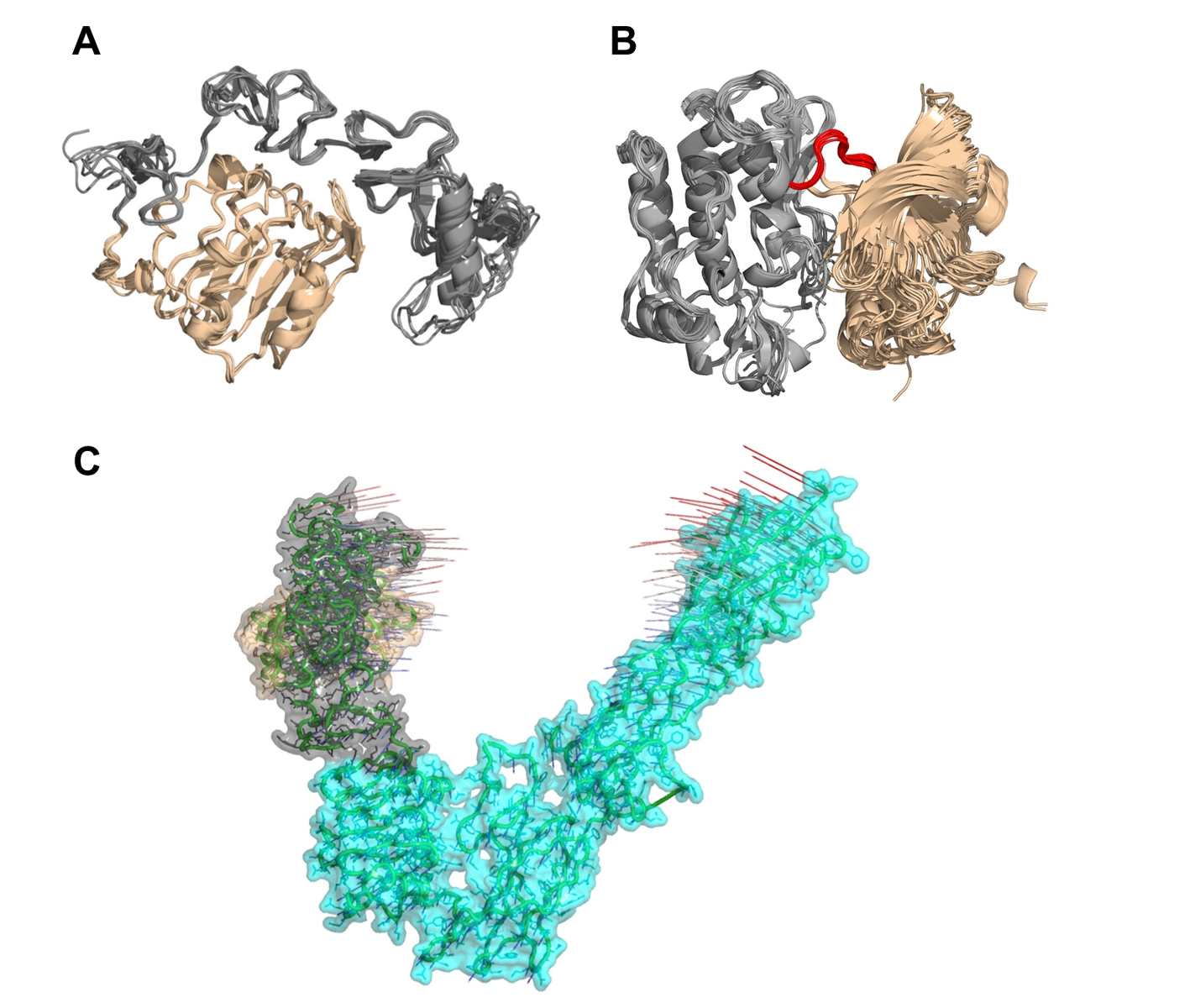 Click for large image | Figure 6. Multiple structural alignments of insulin receptors (IR) to identify the clustroid structure. (A) The experimentally available structures of the L1 domain (wheat) and the cysteine-rich domain (gray) of IR show subtle structural dynamics. (B) It appears that the N-terminal region (wheat) of IR residues Asp1014-Lys1310 undergoes rotation at loop Met1078-Cys1083 (red) with respect to the C-terminal region (gray), resulting in structural dynamics. (C) Normal mode analysis on PDB entry 3LOH_E suggests that the N-terminal L1 domain (wheat) and the CR domain (gray) show a potential for large-scale motions towards each other and the C-terminal of IR ectodomain. |
To better understand the conformational dynamics associated with insulin-IR interaction and to characterize the relative movements of IR domains, quantitative geometric measures across multiple structures of insulin, IR, and their complexes in diverse conditions (e.g. wild-type/mutants, full-length/truncated), can be compared. Examples of geometric measures include the radius of gyration of each insulin binding site on IR, the buried surface areas involved in IR domain-domain interactions, the IR inter-domain hinge angles, and the inter-hinge distances based on the centers of mass of each domain [50]. It seems that the inter-domain flexibility and dynamics of IR serve as the mechanism for the see-saw model of negative cooperativity [51].
The future availability of more IR structures will provide a better understanding of the structural dynamics of IR. For example, advanced techniques such as thermal-factor sharpening can be applied on earlier crystallographic structures, to improve the interpretability of the electron-density maps [15]. At molecular and structural levels, the next challenges include obtaining the full-length structures of IR (through integrative experimental and computational methods), investigating the insulin-stimulated conformational switch of full-length IR, discovering and developing therapeutic insulin mimetics with enhanced receptor binding and favorable pharmacokinetics.
For future study of the insulin-stimulated conformational switch of full-length IR, we recommend the following methods that have successfully been applied on related/other systems: MD simulations [50], harmonic oscillator model to estimate kinetics parameters [19], normal mode analysis (NMA) to determine rotations [52], and multi-scale simulation [53] approach that combines coarse-grained and all-atomistic MD simulations. NMA on the IR ectodomain (PDB: 3LOH_E) suggests that the N-terminal of L1 domain and the CR domain have the potential to move closer to each other (Fig. 6); this motion may be involved in the insulin-stimulated conformational changes of IR. We note that this mouse structure of IR ectodomain, comprising residues Pro31-Ser682 and mutations to stabilize the structure, was resolved in the presence of antibody fragments as crystallization chaperones to aid the structural determination of the IR ectodomain [15]. The antibody fragments serve in minimizing the conformational heterogeneity of IR through locking or clamping the IR ectodomain in a particular subset of conformations [54], whereas we performed the NMA in the absence of the antibody fragments. The integration of multiple methods is essential for obtaining a better picture of the IR mechanisms. Future NMA on the full-length IR is useful to further verify this finding.
| Nascent Nanotechnology-Based Approaches for Controlled Delivery of Insulin | ▴Top |
Conventional subcutaneous administration of insulin has been challenging for diabetics due to poor compliance, local tissue necrosis, pain and nerve damage. To design controlled delivery of insulin, novel nanomaterials, formulations and devices can be identified and optimized, based on the thorough structural understandings of insulin, its heteromerization, and its binding mechanisms to its receptors. Non-invasive administrations of insulin, such as through oral, nasal, pulmonary and transdermal delivery as well as closed-loop smart insulin delivery, are highly desirable.
The closed-loop smart insulin delivery has the potential to mimic the pancreatic function. Because of the controlled regulation of insulin release rate from biodegradable polymeric biomaterials [55], the benefits of the closed-loop smart insulin delivery include minimal blood glucose fluctuation and reduced hypoglycemia risk. This delivery is also more resistant to enzymatic degradation, denaturation and immune recognition, which are major problems faced by insulin delivered through non-invasive administration. To design closed-loop smart insulin delivery, insulin is embedded in a polymeric matrix that contains glucose-sensitive molecules, such as phenylboronic acid [56], glucose oxidase [55], and glucose binding proteins, mainly lectins [57] such as concanavalin A that possesses four glucose binding sites.
| Conclusion | ▴Top |
In this review, we have summarized the progress of structural biology of insulin and its receptor hitherto, and proposed testable hypotheses that can propel our knowledge on the structural dynamics of insulin and its receptor. Literature mining of insulin and its receptor implies their functional relationships with other hormones, highlighting the importance of inter-molecular cross-talks. We envisage that increasing availability of structural data from both experimental and computational methods, together with their synergistic integration, will be helpful to refine all-atoms models of full-length IR and its complex with insulin and other growth factors. Thorough understandings of their molecular structures, functions, structural dynamics and mechanisms, are critical in therapeutic designs for treating diabetes.
Acknowledgement
We thank our colleagues at Khalifa University for critically reading our manuscript and acknowledge the 2015 Khalifa University Internal Research Fund Level 1 210070 for funding our research.
Conflict of Interest
We declare no conflict of interest.
Abbreviations
CR: cysteine-rich domain; Fn: fibronectin type III domains; Kd: dissociation constant; L1: large domain 1 of insulin receptor (leucine-rich repeats); L2: large domain 2 of insulin receptor (leucine-rich repeats); MD: molecular dynamics; SH2: Src homology 2; TK: tyrosine kinase domain; TM: transmembrane domain
| References | ▴Top |
- Yang Y, Hua QX, Liu J, Shimizu EH, Choquette MH, Mackin RB, Weiss MA. Solution structure of proinsulin: connecting domain flexibility and prohormone processing. J Biol Chem. 2010;285(11):7847-7851.
doi pubmed - Huang K, Dong J, Phillips NB, Carey PR, Weiss MA. Proinsulin is refractory to protein fibrillation: topological protection of a precursor protein from cross-beta assembly. J Biol Chem. 2005;280(51):42345-42355.
doi pubmed - Wang F, Hull RL, Vidal J, Cnop M, Kahn SE. Islet amyloid develops diffusely throughout the pancreas before becoming severe and replacing endocrine cells. Diabetes. 2001;50(11):2514-2520.
doi pubmed - Menting JG, Whittaker J, Margetts MB, Whittaker LJ, Kong GK, Smith BJ, Watson CJ, et al. How insulin engages its primary binding site on the insulin receptor. Nature. 2013;493(7431):241-245.
doi pubmed - Chang X, Jorgensen AM, Bardrum P, Led JJ. Solution structures of the R6 human insulin hexamer. Biochemistry. 1997;36(31):9409-9422.
doi pubmed - De Meyts P. Insulin and its receptor: structure, function and evolution. Bioessays. 2004;26(12):1351-1362.
doi pubmed - Tidor B, Karplus M. The contribution of vibrational entropy to molecular association. The dimerization of insulin. J Mol Biol. 1994;238(3):405-414.
doi pubmed - De Meyts P, Whittaker J. Structural biology of insulin and IGF1 receptors: implications for drug design. Nat Rev Drug Discov. 2002;1(10):769-783.
doi pubmed - McKern NM, Lawrence MC, Streltsov VA, Lou MZ, Adams TE, Lovrecz GO, Elleman TC, et al. Structure of the insulin receptor ectodomain reveals a folded-over conformation. Nature. 2006;443(7108):218-221.
doi pubmed - Sparrow LG, McKern NM, Gorman JJ, Strike PM, Robinson CP, Bentley JD, Ward CW. The disulfide bonds in the C-terminal domains of the human insulin receptor ectodomain. J Biol Chem. 1997;272(47):29460-29467.
doi pubmed - Schaffer L, Ljungqvist L. Identification of a disulfide bridge connecting the alpha-subunits of the extracellular domain of the insulin receptor. Biochem Biophys Res Commun. 1992;189(2):650-653.
doi - Ren S, Yang G, He Y, Wang Y, Li Y, Chen Z. The conservation pattern of short linear motifs is highly correlated with the function of interacting protein domains. BMC Genomics. 2008;9:452.
doi pubmed - Hubbard SR. The insulin receptor: both a prototypical and atypical receptor tyrosine kinase. Cold Spring Harb Perspect Biol. 2013;5(3):a008946.
doi pubmed - Hubbard SR. Structural biology: Insulin meets its receptor. Nature. 2013;493(7431):171-172.
doi pubmed - Smith BJ, Huang K, Kong G, Chan SJ, Nakagawa S, Menting JG, Hu SQ, et al. Structural resolution of a tandem hormone-binding element in the insulin receptor and its implications for design of peptide agonists. Proc Natl Acad Sci U S A. 2010;107(15):6771-6776.
doi pubmed - Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117-1134.
doi pubmed - Trabuco LG, Villa E, Mitra K, Frank J, Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16(5):673-683.
doi pubmed - Vashisth H. Flexibility in the insulin receptor ectodomain enables docking of insulin in crystallographic conformation observed in a hormone-bound microreceptor. Membranes (Basel). 2014;4(4):730-746.
doi pubmed - Kiselyov VV, Versteyhe S, Gauguin L, De Meyts P. Harmonic oscillator model of the insulin and IGF1 receptors' allosteric binding and activation. Mol Syst Biol. 2009;5:243.
doi pubmed - Ward CW, Lawrence MC. Ligand-induced activation of the insulin receptor: a multi-step process involving structural changes in both the ligand and the receptor. Bioessays. 2009;31(4):422-434.
doi pubmed - De Meyts P. The insulin receptor: a prototype for dimeric, allosteric membrane receptors? Trends Biochem Sci. 2008;33(8):376-384.
doi pubmed - Thorsoe KS, Schlein M, Steensgaard DB, Brandt J, Schluckebier G, Naver H. Kinetic evidence for the sequential association of insulin binding sites 1 and 2 to the insulin receptor and the influence of receptor isoform. Biochemistry. 2010;49(29):6234-6246.
doi pubmed - Lukman S, Grant BJ, Gorfe AA, Grant GH, McCammon JA. The distinct conformational dynamics of K-Ras and H-Ras A59G. PLoS Comput Biol. 2010;6(9).
doi pubmed - Menting JG, Yang Y, Chan SJ, Phillips NB, Smith BJ, Whittaker J, Wickramasinghe NP, et al. Protective hinge in insulin opens to enable its receptor engagement. Proc Natl Acad Sci U S A. 2014;111(33):E3395-3404.
doi pubmed - Shoelson S, Haneda M, Blix P, Nanjo A, Sanke T, Inouye K, Steiner D, et al. Three mutant insulins in man. Nature. 1983;302(5908):540-543.
doi pubmed - Hua QX, Shoelson SE, Inouye K, Weiss MA. Paradoxical structure and function in a mutant human insulin associated with diabetes mellitus. Proc Natl Acad Sci U S A. 1993;90(2):582-586.
doi pubmed - Zoete V, Meuwly M, Karplus M. A comparison of the dynamic behavior of monomeric and dimeric insulin shows structural rearrangements in the active monomer. J Mol Biol. 2004;342(3):913-929.
doi pubmed - Li Q, Wong YL, Kang C. Solution structure of the transmembrane domain of the insulin receptor in detergent micelles. Biochim Biophys Acta. 2014;1838(5):1313-1321.
doi pubmed - Lee J, Miyazaki M, Romeo GR, Shoelson SE. Insulin receptor activation with transmembrane domain ligands. J Biol Chem. 2014;289(28):19769-19777.
doi pubmed - Hubbard SR, Miller WT. Receptor tyrosine kinases: mechanisms of activation and signaling. Curr Opin Cell Biol. 2007;19(2):117-123.
doi pubmed - Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539.
doi pubmed - Mathews ST, Chellam N, Srinivas PR, Cintron VJ, Leon MA, Goustin AS, Grunberger G. Alpha2-HSG, a specific inhibitor of insulin receptor autophosphorylation, interacts with the insulin receptor. Mol Cell Endocrinol. 2000;164(1-2):87-98.
doi - Goustin AS, Abou-Samra AB. The "thrifty" gene encoding Ahsg/Fetuin-A meets the insulin receptor: Insights into the mechanism of insulin resistance. Cell Signal. 2011;23(6):980-990.
doi pubmed - Kim JJ, Accili D. Signalling through IGF-I and insulin receptors: where is the specificity? Growth Horm IGF Res. 2002;12(2):84-90.
doi - Kholodenko BN. Untangling the signalling wires. Nat Cell Biol. 2007;9(3):247-249.
doi pubmed - Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A, Goldfine ID, et al. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol. 1999;19(5):3278-3288.
pubmed - Denley A, Wallace JC, Cosgrove LJ, Forbes BE. The insulin receptor isoform exon 11- (IR-A) in cancer and other diseases: a review. Horm Metab Res. 2003;35(11-12):778-785.
pubmed - Sciacca L, Vigneri R, Tumminia A, Frasca F, Squatrito S, Frittitta L, Vigneri P. Clinical and molecular mechanisms favoring cancer initiation and progression in diabetic patients. Nutr Metab Cardiovasc Dis. 2013;23(9):808-815.
doi pubmed - Soluble insulin receptor ectodomain is elevated in the plasma of patients with diabetes. Diabetes. 2007;56(8):2028-2035.
doi pubmed - Yuasa T, Amo K, Ishikura S, Nagaya H, Uchiyama K, Hashida S, Ebina Y. Development of in vitro model of insulin receptor cleavage induced by high glucose in HepG2 cells. Biochem Biophys Res Commun. 2014;445(1):236-243.
doi pubmed - Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond). 2012;122(6):253-270.
doi pubmed - Rena G, Pearson ER, Sakamoto K. Molecular mechanism of action of metformin: old or new insights? Diabetologia. 2013;56(9):1898-1906.
doi pubmed - Longo N, Wang Y, Smith SA, Langley SD, DiMeglio LA, Giannella-Neto D. Genotype-phenotype correlation in inherited severe insulin resistance. Hum Mol Genet. 2002;11(12):1465-1475.
doi pubmed - Lukman S, Lane DP, Verma CS. Mapping the structural and dynamical features of multiple p53 DNA binding domains: insights into loop 1 intrinsic dynamics. PLoS One. 2013;8(11):e80221.
doi pubmed - Lukman S, Grant GH, Bui JM. Unraveling evolutionary constraints: a heterogeneous conservation in dynamics of the titin Ig domains. FEBS Lett. 2010;584(6):1235-1239.
doi pubmed - Sim K, Yap G-E, Hardoon DR, Gopalkrishnan V, Cong G, Lukman S. Centroid-Based Actionable 3D Subspace Clustering. IEEE Trans Knowl Data Eng. 2013;25:1213-1226.
doi - Lukman S, Verma CS, Fuentes G. Exploiting Protein Intrinsic Flexibility in Drug Design. In: Han K-L, Zhang X, Yang M-J, editors. Protein Conformational Dynamics, vol. 805, Cham: Springer International Publishing; 2014, p. 245-269.
doi pubmed - Schames JR, Henchman RH, Siegel JS, Sotriffer CA, Ni H, McCammon JA. Discovery of a novel binding trench in HIV integrase. J Med Chem. 2004;47(8):1879-1881.
doi pubmed - Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, Fiore F, et al. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J Med Chem. 2008;51(18):5843-5855.
doi pubmed - Vashisth H. Theoretical and computational studies of peptides and receptors of the insulin family. Membranes (Basel). 2015;5(1):48-83.
doi - Vashisth H, Abrams CF. Docking of insulin to a structurally equilibrated insulin receptor ectodomain. Proteins. 2010;78(6):1531-1543.
doi - Lukman S, Grant GH. A network of dynamically conserved residues deciphers the motions of maltose transporter. Proteins. 2009;76(3):588-597.
doi pubmed - Samiotakis A, Homouz D, Cheung MS. Multiscale investigation of chemical interference in proteins. J Chem Phys. 2010;132(17):175101.
doi pubmed - Griffin L, Lawson A. Antibody fragments as tools in crystallography. Clin Exp Immunol. 2011;165(3):285-291.
doi pubmed - Gu Z, Aimetti AA, Wang Q, Dang TT, Zhang Y, Veiseh O, Cheng H, et al. Injectable nano-network for glucose-mediated insulin delivery. ACS Nano. 2013;7(5):4194-4201.
doi pubmed - Ma R, Yang H, Li Z, Liu G, Sun X, Liu X, An Y, et al. Phenylboronic acid-based complex micelles with enhanced glucose-responsiveness at physiological pH by complexation with glycopolymer. Biomacromolecules. 2012;13(10):3409-3417.
doi pubmed - Wu S, Huang X, Du X. Glucose- and pH-responsive controlled release of cargo from protein-gated carbohydrate-functionalized mesoporous silica nanocontainers. Angew Chem Int Ed Engl. 2013;52(21):5580-5584.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.